OLIGODENDROGLIOMA
Luigi Cuccurullo
|
L’oligodendroglioma è un tumore di natura gliale, localizzato quasi sempre a livello degli emisferi cerebrali; esso è costituito da cellule simili ad oligodendrociti e caratterizzate a livello genetico-molecolare di delezione del 1p e 19q (W.H.O.).
L’oligodendroglioma si localizza con elevata frequenza a livello della corteccia cerebrale e/o della sostanza bianca degli emisferi cerebrali; le aree maggiormente coinvolte sono in ordine decrescente, i lobi frontali, quelli temporali, quindi quelli occipitali; sono riportati in letteratura casi di oligodendromi indovati a livello del cervelletto, del ponte, dei nuclei delle fosse, del midollo spinale.
Sono descritti casi in cui i focolai di oligodendromi sono duplici e situati spazialmente simili a livello di due emisferi.
La neoplasia è localizzata in sede paraventricolare accrescendosi può aprirsi in una cavità ventricolare per poi diffondersi per via liquorale.
Caratteri macroscopici
L’oligodendroglioma (grado II) appare quale neofromazione apparentemente ben delimitata, di consistenza morbido-molliccia, abitualmente di colore grigio-rossastro.
Questi caratteri di base possono modificarsi nel corso del suo accrescersi, per il subentrare di fenomeni secondari; così, può acquisire un aspetto gelatinoso per il manifestarsi di processi di degenerazione mucoide, oppure acquisire un colore rosso per iperemia e/o emorragia oppure un colore bianco-latte per una marcata, diffusa condizione di edema.
Al taglio la neoplasia può apparire uniforme e compatta oppure lasciar evidenziare micro-macro escavazioni ricolme di liquido mucinoso oppure siero-emorragico. Al taglio, il coletto può trovare resistenza con stridore per la presena di microfori calcifici oppure rari piccoli aggregati metaplastici, osteoidi. La crescita dell’oligodendroglioma è di tipo infiltrativo, pertanto si ha sempre un margine visibile a occhio nudo e un altro identificabile al microscopio luce.
Nel suo accrescersi può superare la corteccia cerebrale, occupare gli spazi sub-aracnoidei, infiltrare le leptomeningi, raggiungere gli spazi sub-durali ed attecchire a livello degli strati interni della teca cranuica; allo stesso modo, se il focolaio di origine è nella sostanza bianca, il tumore può proiettarsi ed aprirsi nella cavità ventricolare per poi diffondersi per via liquorale. L’oligodendroglioma grado III (oligodendroglioma anaplastico) presenta i medesimi caratteri macroscopici con variazioni in senso peggiorativo. La topografia della neoplasia è simile a quella dianzi descritta; la consistenza è estremamente ridotta per la presenza di aree di necrosi e di emorragie; il colore è variegato per la co-esistenza di diversi processi regressivi e necrotico-emorragici; le superfici di taglio mostrano numerose escavazioni con margini anfrattuosi ricolme di materiale semifluido; la crescita, di tipo infiltrativo è rapida e quasi sempre ha caratteri destruenti le strutture pre-esistenti.
CARATTERI ISTOPATOLOGICI
L’oligodendroglioma grado III si rivela al microscopio luce essere una neoplasia a cellularità medio-alta con livelli di densità variabili da zona a zona nell’ambito dello stesso caso. Le cellule si dispongono spazialmente in modo diffuso oppure formano travate, cordoni o filiere monostratificate variamente orientate.
Questi elementi sono quasi sempre monomorfi, hanno forma rotondeggiante, hanno un esiguo citoplasma con corti prolungamenti e sono occupati da un nucleo sferoidale, normo-cromatinico, apparentemente privo di nucleolo. Un aspetto caratterizzante è dato dalla esistenza di un alone iperchiaro perinucleare.
Fig.1 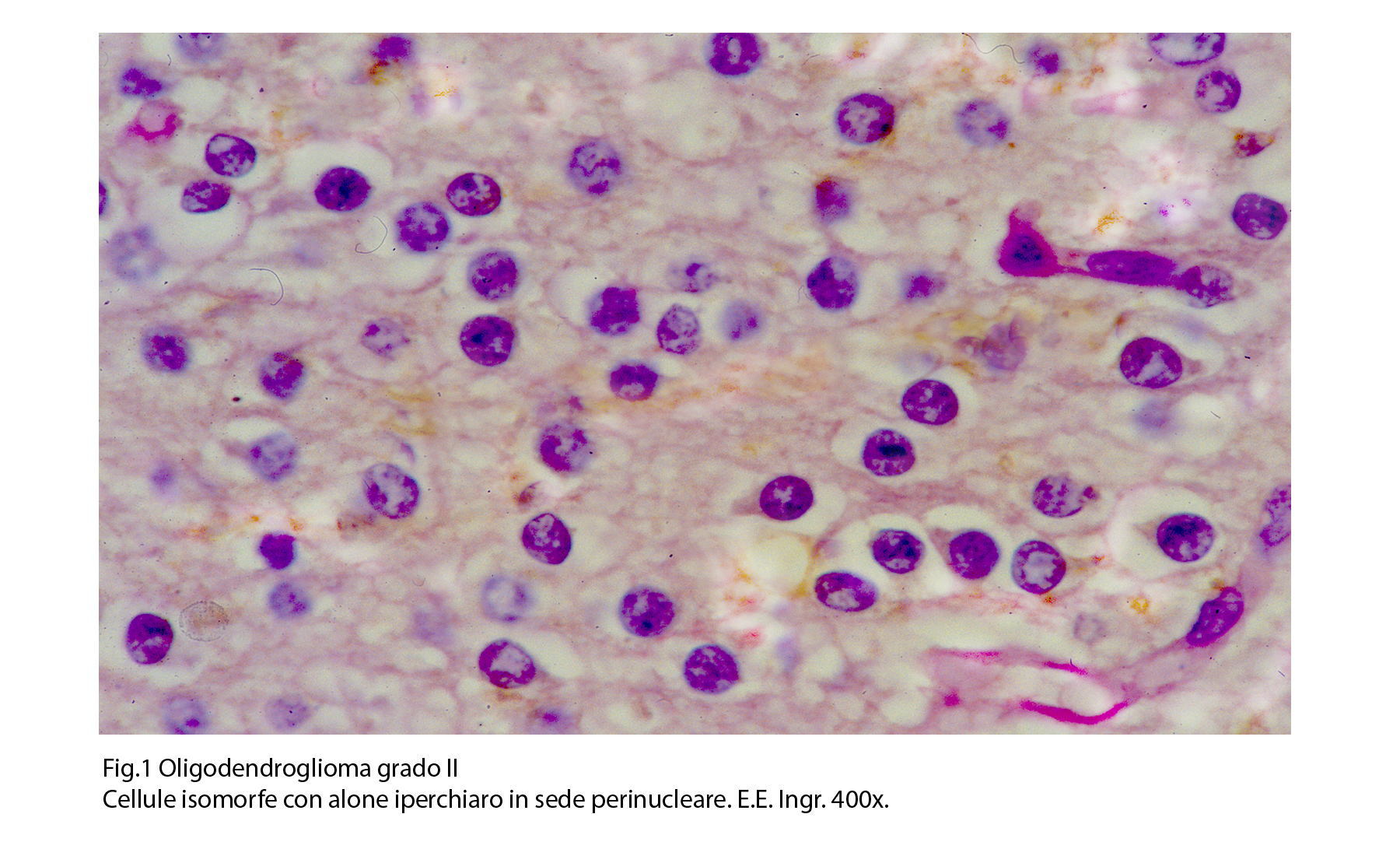 Fig.2
Fig.2 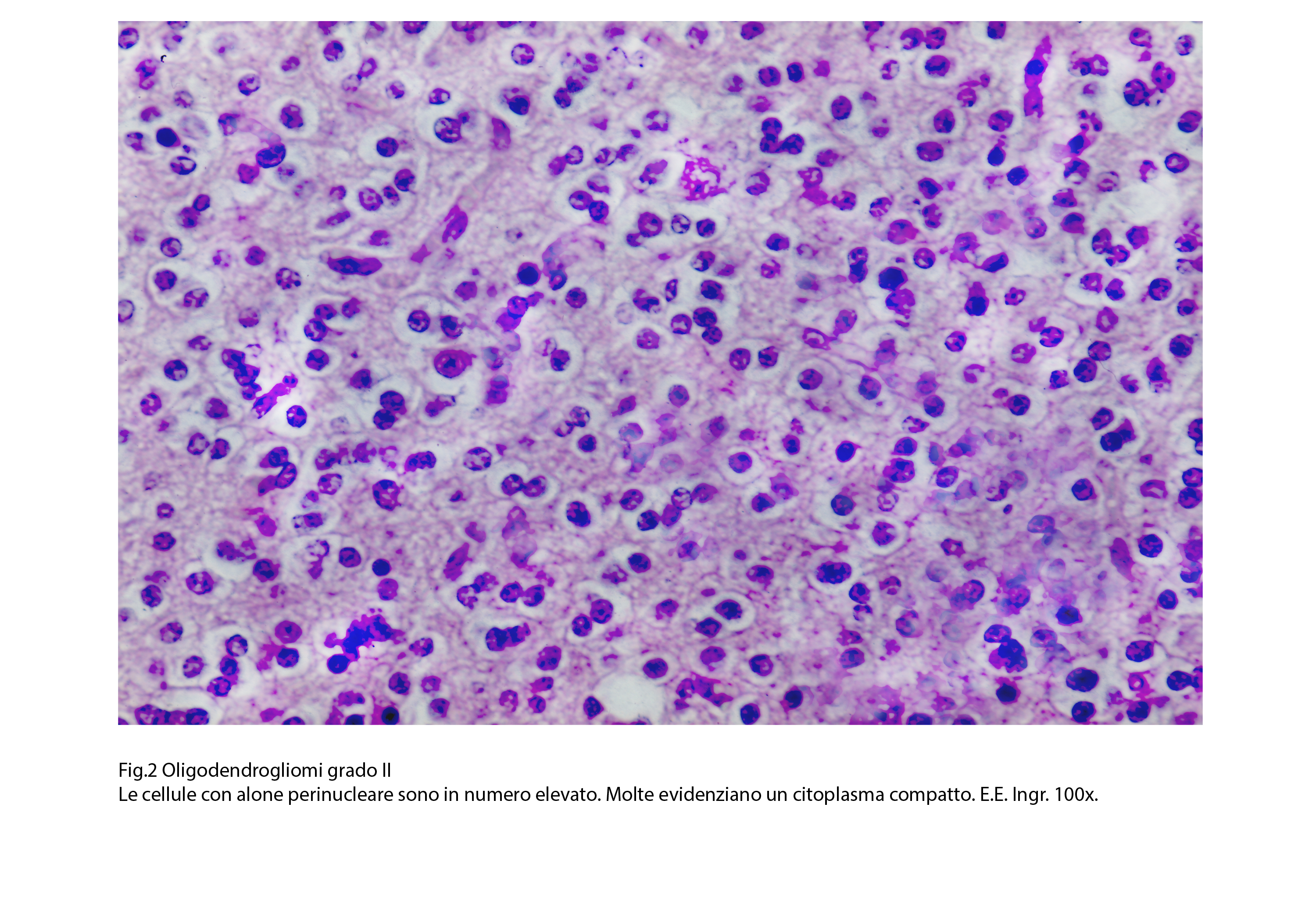 Fig.3
Fig.3 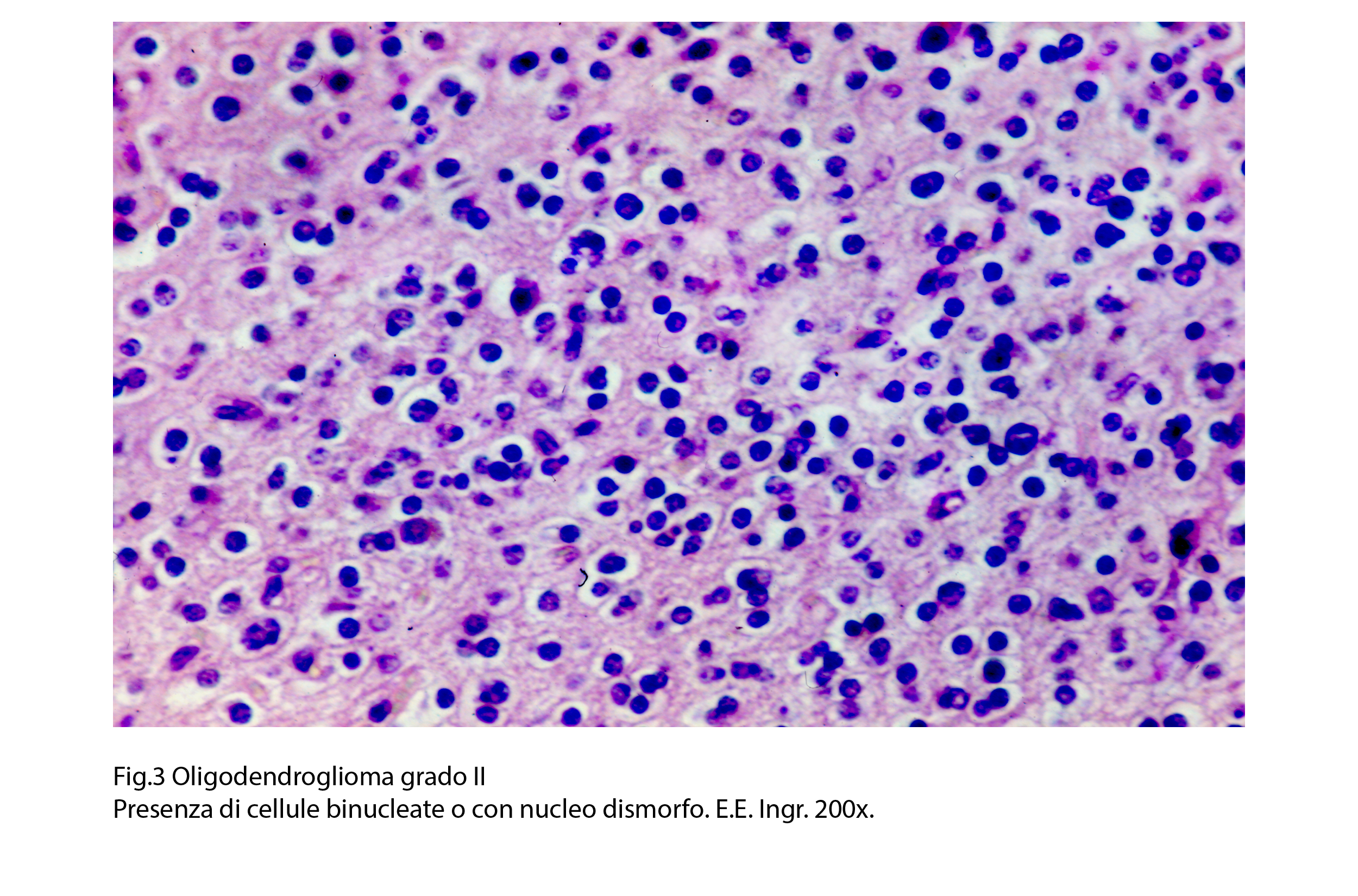
Il citoplasma di questi elementi può apparire punteggiato, in modo irregolare da grandi acidofili.
Fig.4 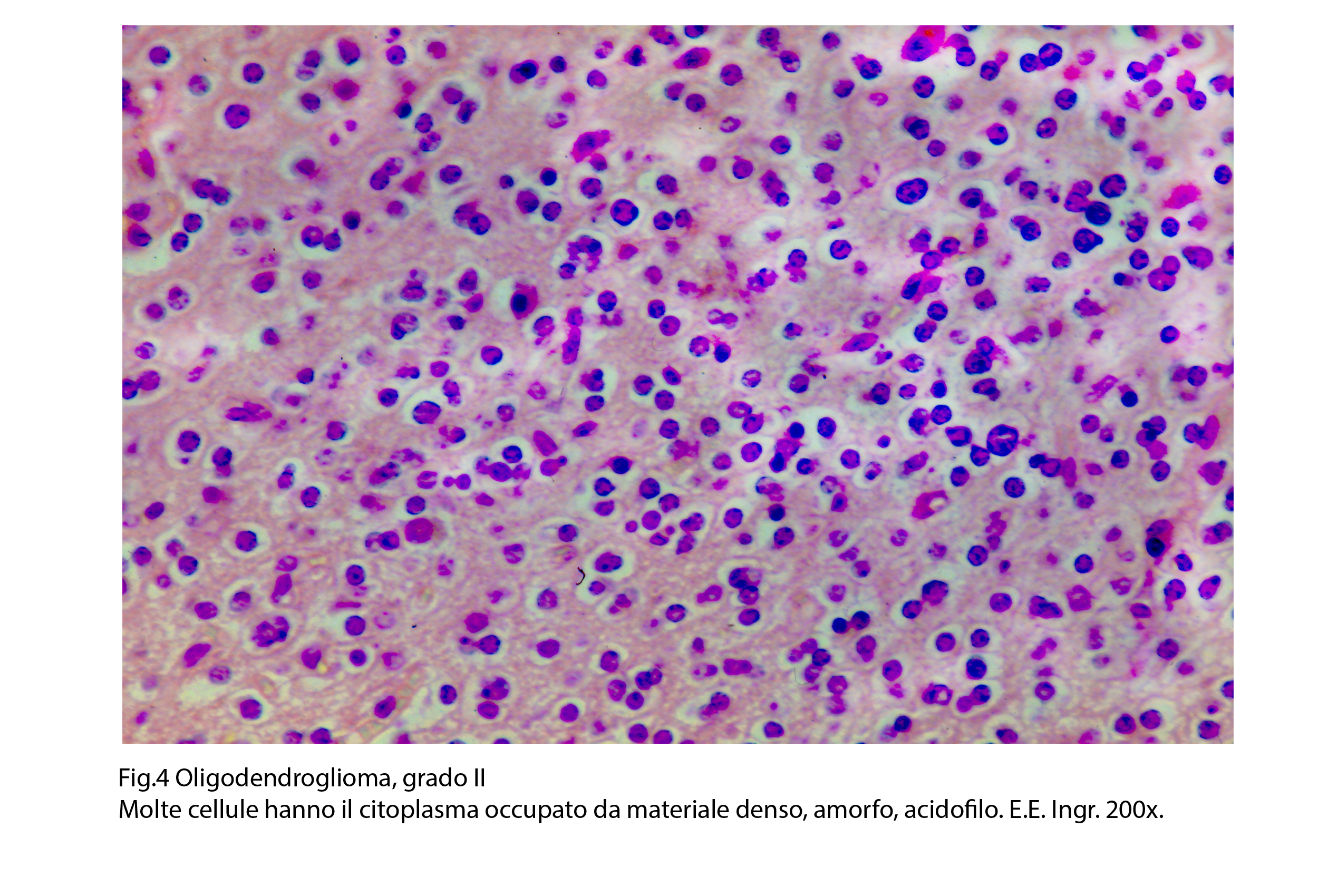
Sono descritti casi di oligodendroglioma, i quali sono forniti di nuclei irregolari, polimorfi, fusati, ipercromatinici ma privi di aggressività biologica e/o clinica.
Fig.5 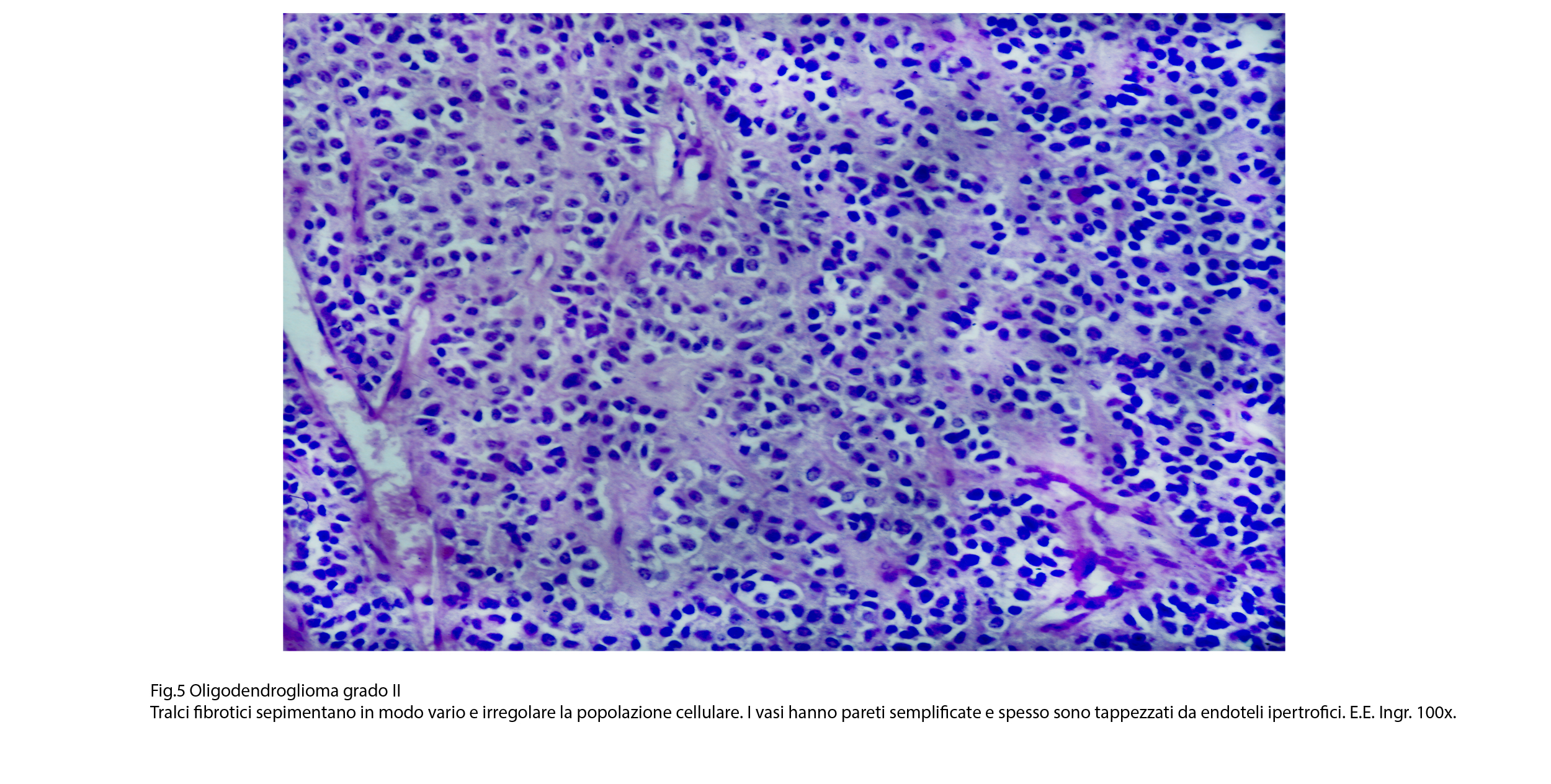 Fig.6
Fig.6 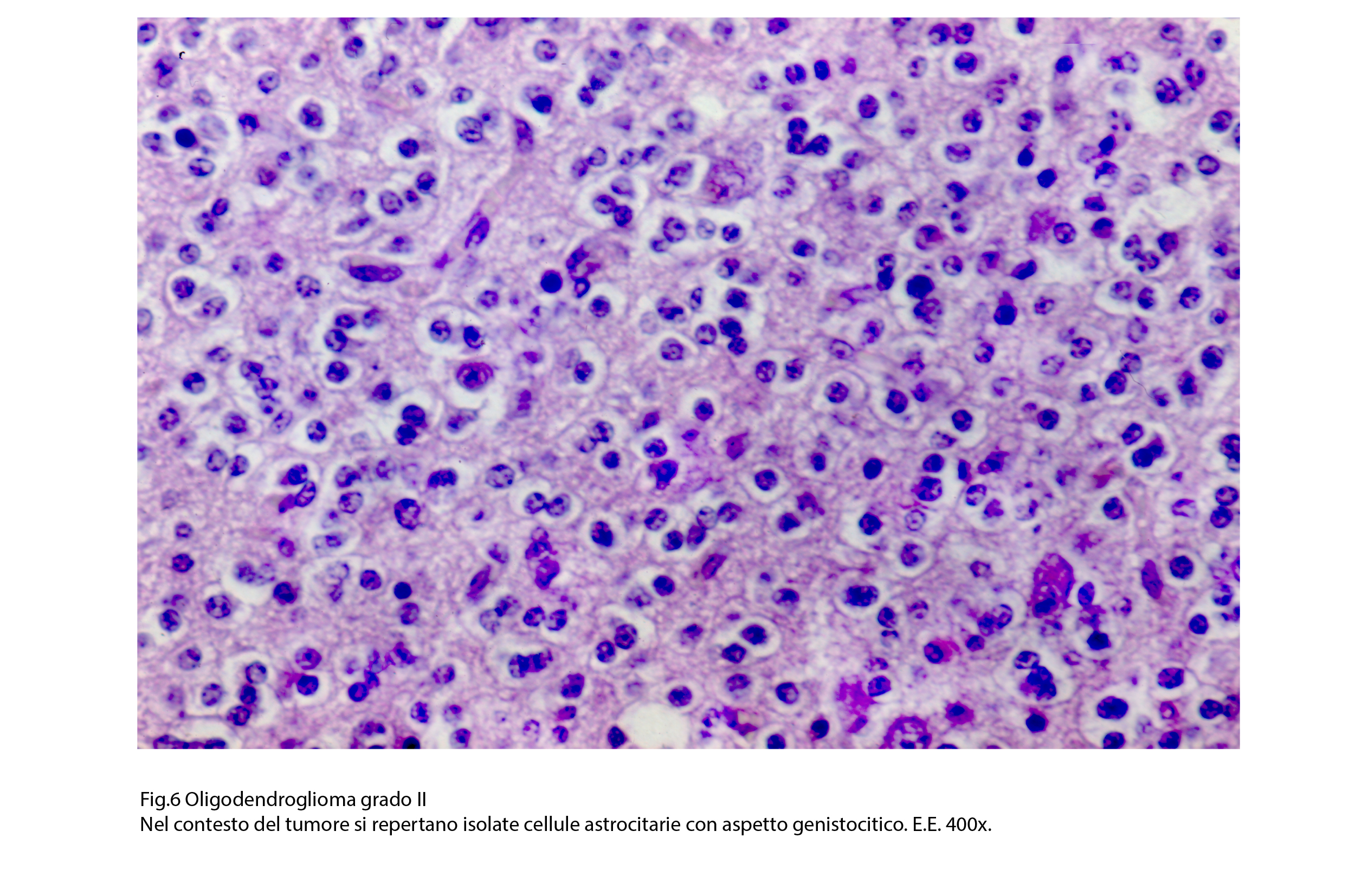
Qualunque sia la forma dei nuclei, nei glioblastomi le mitosi tipiche o atipiche sono poco frequenti.
Nel contesto della popolazione dell’oligodendroglioma si possono riconoscere elementi astrocitari di vario tipo e in particolare di tipo genistocitico; questi sono considerati come cellule reattive oppure come elementi rimasti intrappolati nel contesto del tumore. E’ frequente il riscontro di immagini di satellitosi; esse consistono nella disposizione delle cellule neoplastiche ad avvolgere un neurone o un piccolo vaso. Le cellule dell’oligodendroglioma possono essere sede di rigonfiamento idropilo e di conseguenza acquisiscono un aspetto globoso e diafano, con membrana cellulare ben demarcata.
Fig.7 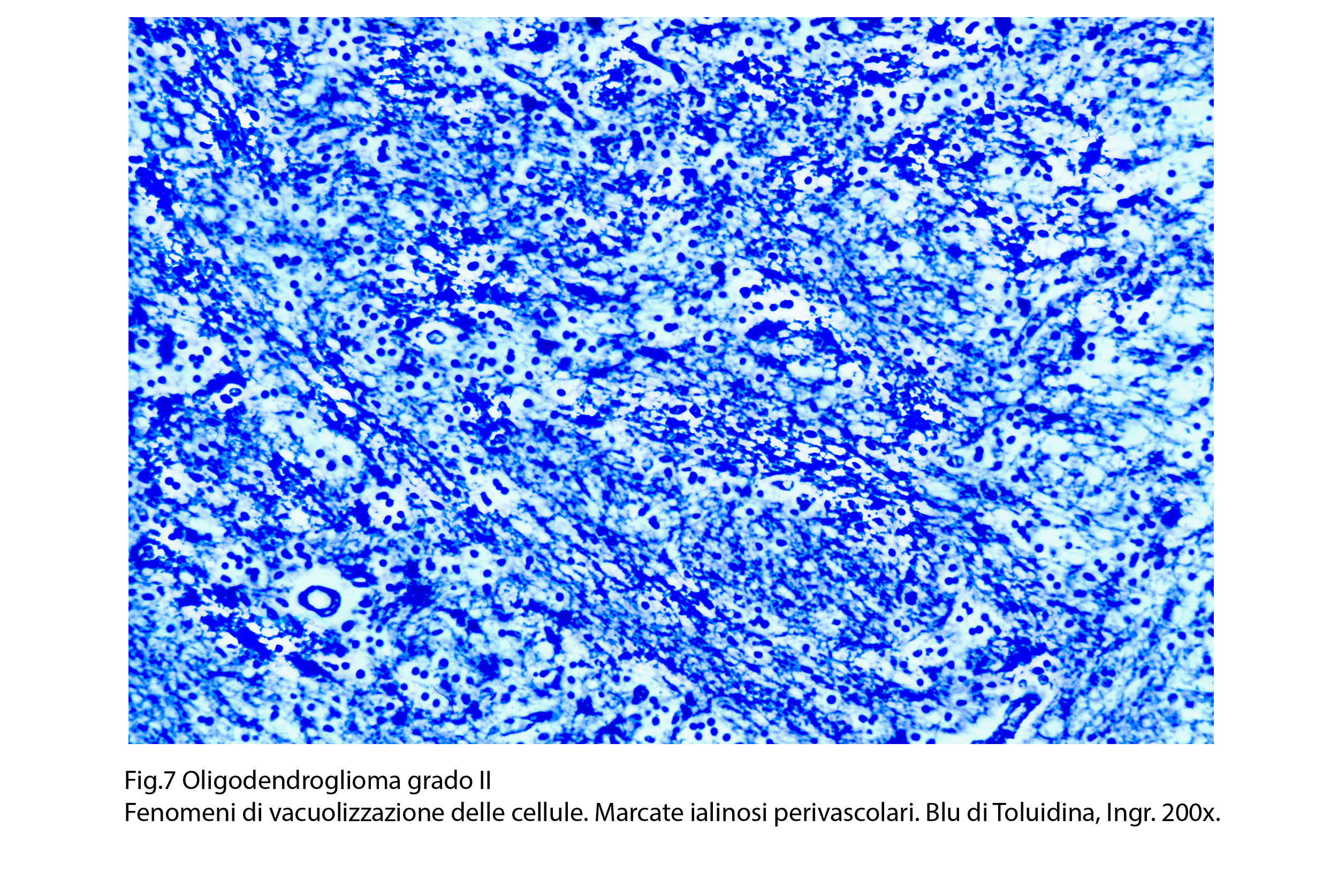
Le cavità micro-macroscopiche sono prive di pareti proprie; esse sono delimitate da strati di elementi neoplastici e il loro spazio cavo contiene ectonucine siero-eritrocitario. La quota vascolare è ben sviluppata ed è costituita prevalentemente da capillari in proliferazione; essa è distribuita in modo asimmetrico o altamente irregolare. Le pareti di questi vasi sono semplificate, sottili, sono rivestiti da endoteli alti, ipertrofici e sono spesso avvolte da elementi linfo-monocitari o da cellule neoplastiche (satellitosi). La componente stromale è data da fibre collagene che si dispongono a tralci, i quali sepimentano irregolarmente la neoplasia; da questi tralci si dipartono fibre reticolari precollagene che sono organizzate in modo da avvolgere piccoli gruppi di cellule eoplastiche e capillari.
Fig.8 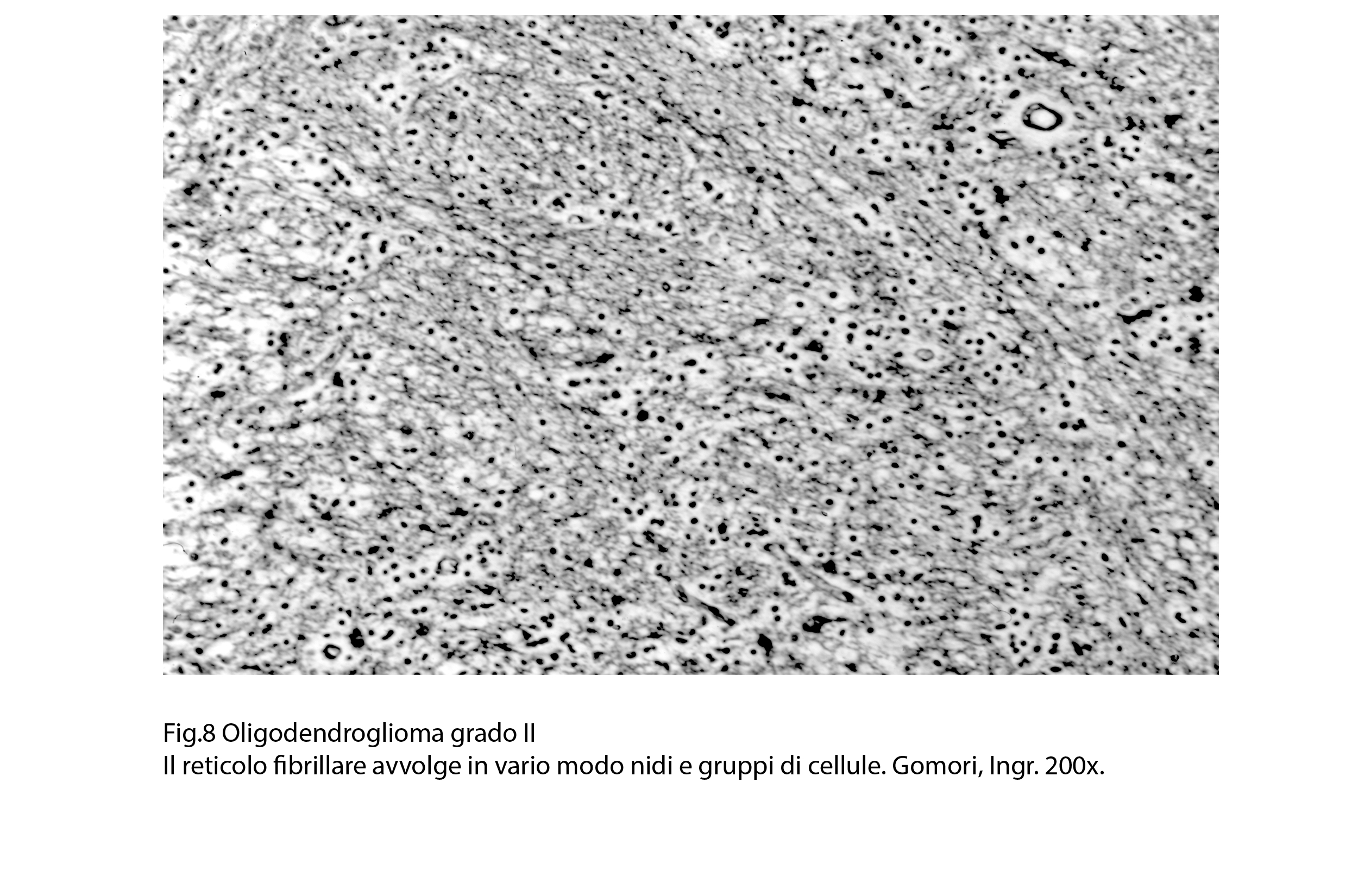
Molto frequentemente si riscontrano negli oligodendrogliomi piccoli depositi di Sali di calcio; questi si raccolgono negli spazi intercellulari, in sede peivascolare o nel contesto delle tonache media e avventizia dei vasi.
Fig.9 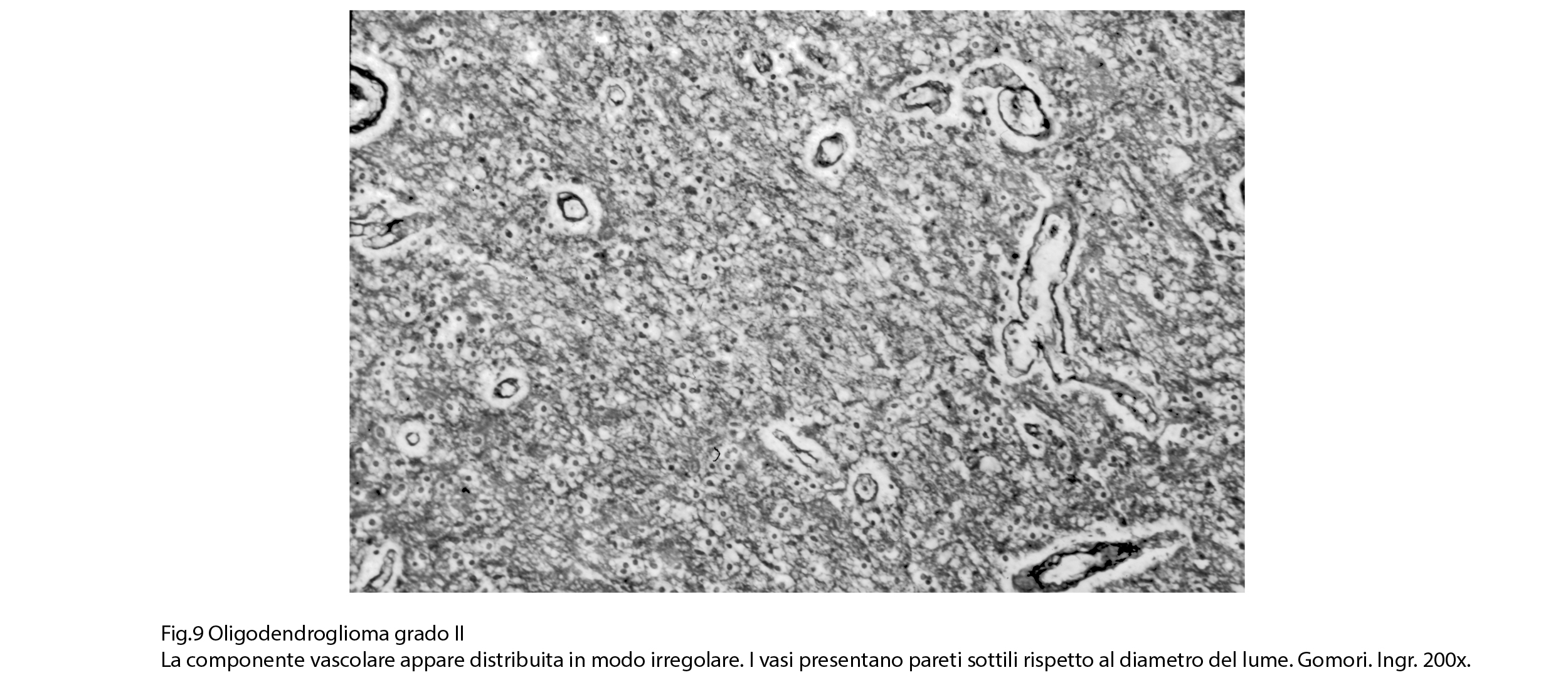
Sono stati segnalati in letteratura casi di oligodendrogliomi contenenti calcosferi o, per metaplasia, tessuto osteroide. Spesso, a livello della componente vasculo-stromale ai fenomeni di calcificazione si possono associare processi di ialinosi che inducono una riduzione del lume vascolare.
I caratteri ultrastrutturali degli oligodendrogliomi grado II sottolineano con dettagli caratteri citologici già documentati mediante la microspia luce. Il nucleo appare normocromatinico, ed è delimitato da una netta membrana basale. Il citoplasma è sede di quote ben rappresentate di organuli, tra una netta membrana cellulare ed è sede di piccole e corte sporgenze del citoplasma. L’alone perinucleare appare diafano, trasparente ed è privo di contenuto visibile agli elettroni.
Fig.10 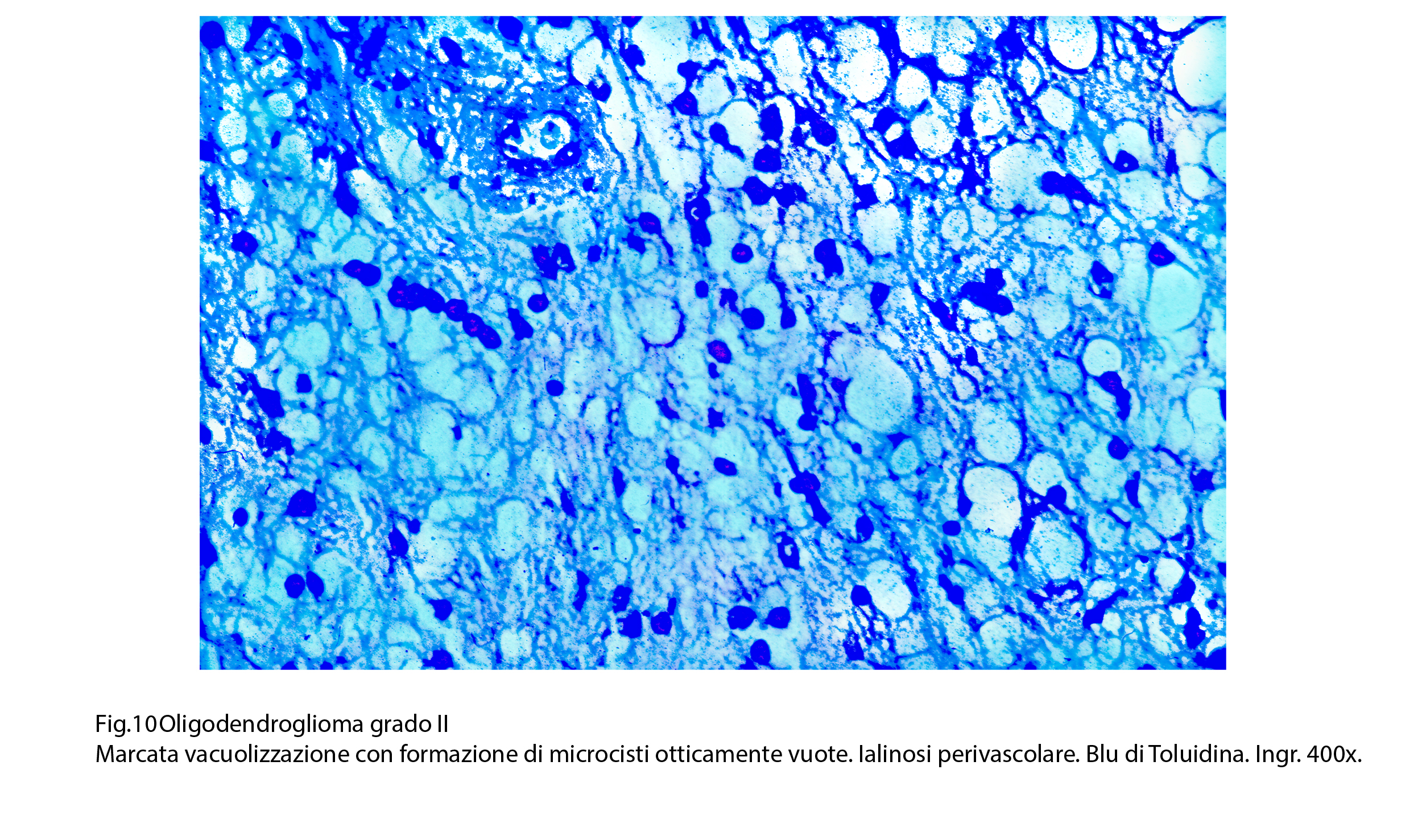
Le cellule tra loro sono adese senza strutture giunzionali, pertanto spesso si formano ampi spazi intercellulari. Le cellule possono subire processi regressivi quali il rigonfiamento idrofilo o addirittura processi di necrobiosi con frammentazione della membrana cellulare e disorganizzazione del citoplasma.
Fig.11 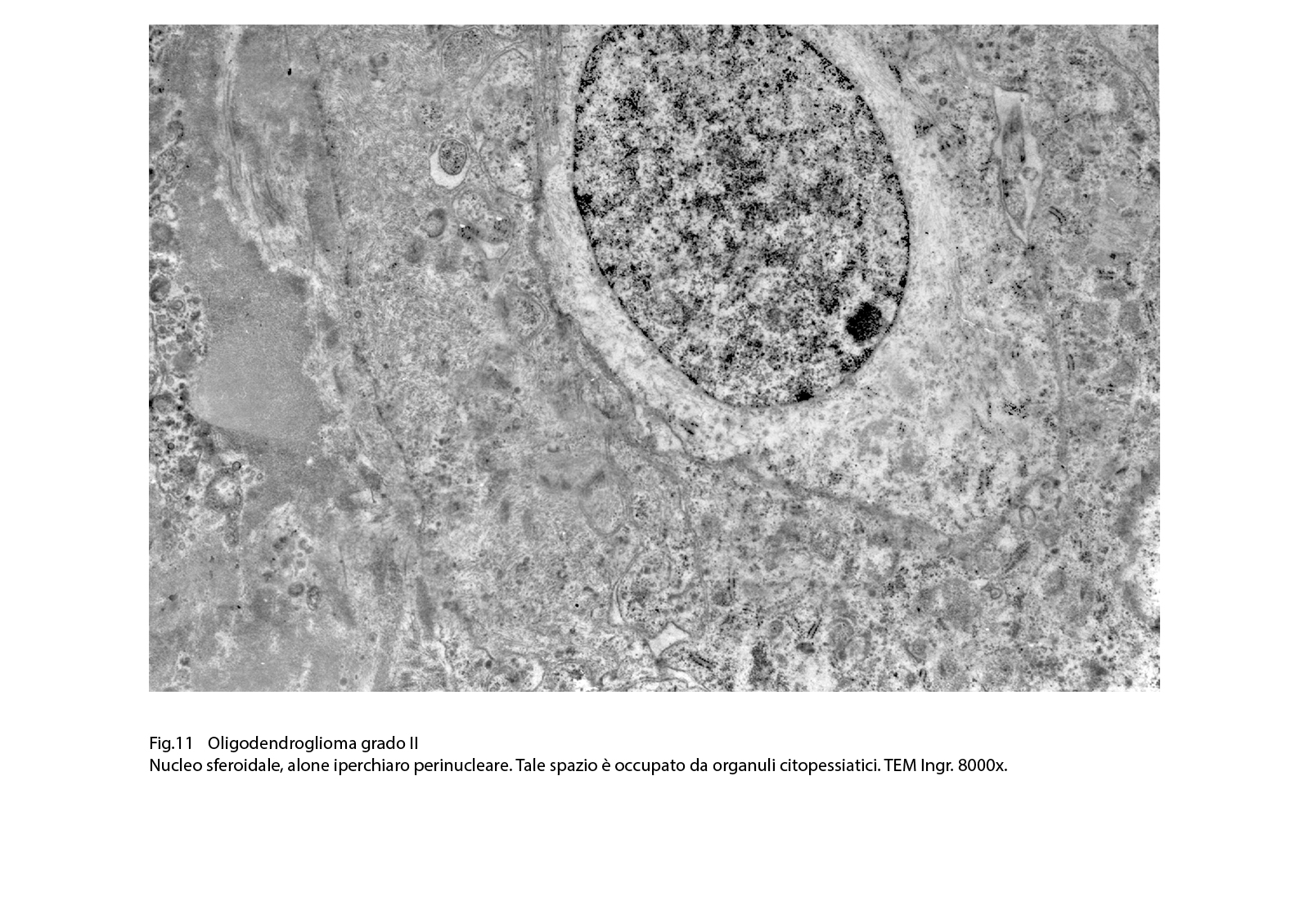 Fig.12
Fig.12 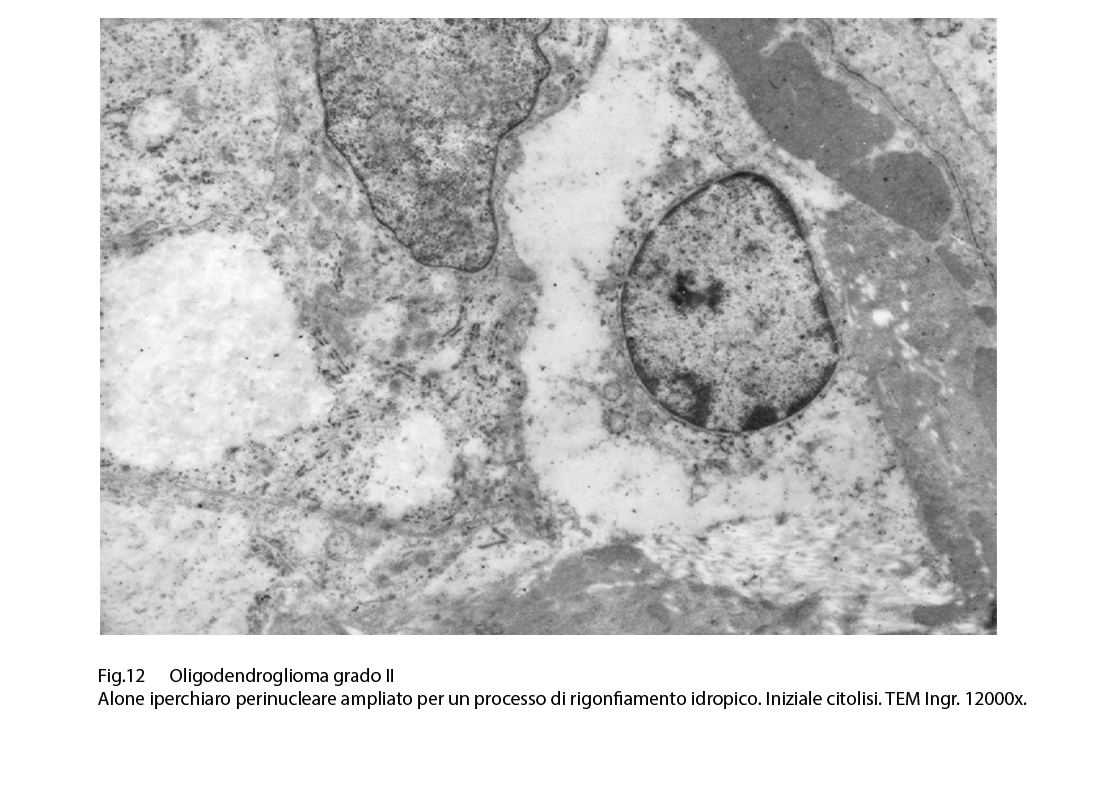
Immunoistochimica
Il profilo immunoistochimico degli oligodendrogliomi grado II è così articolato:
GFAP = negativo
S-100 = positivo
Proteina mielina basica (MBP) = positiva
Proteina proteolipidica (PCP) = positiva
Glicoproteina associata a mielina = (MAG) = positiva
OLIG ½ = positivo
MAP2 = positiva
SOx10= positiva
Vimentina = positività infrequente
W.H.O., 2007
Fig.13 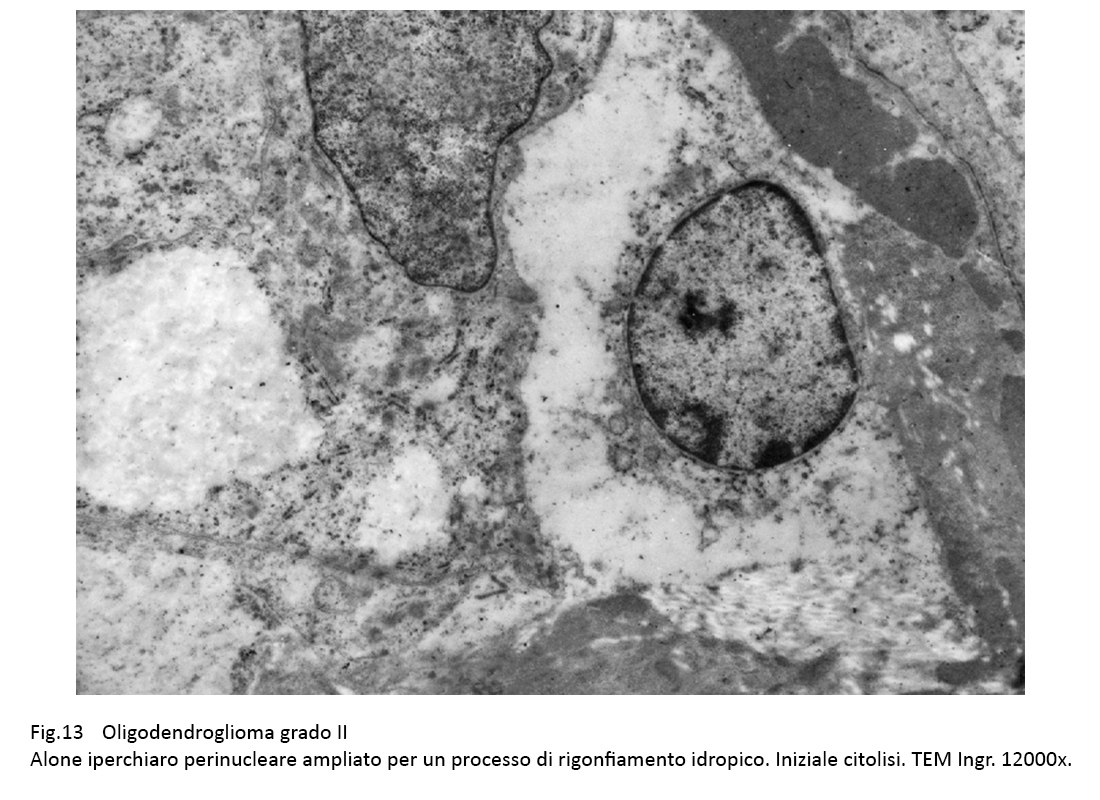 Fig.14
Fig.14 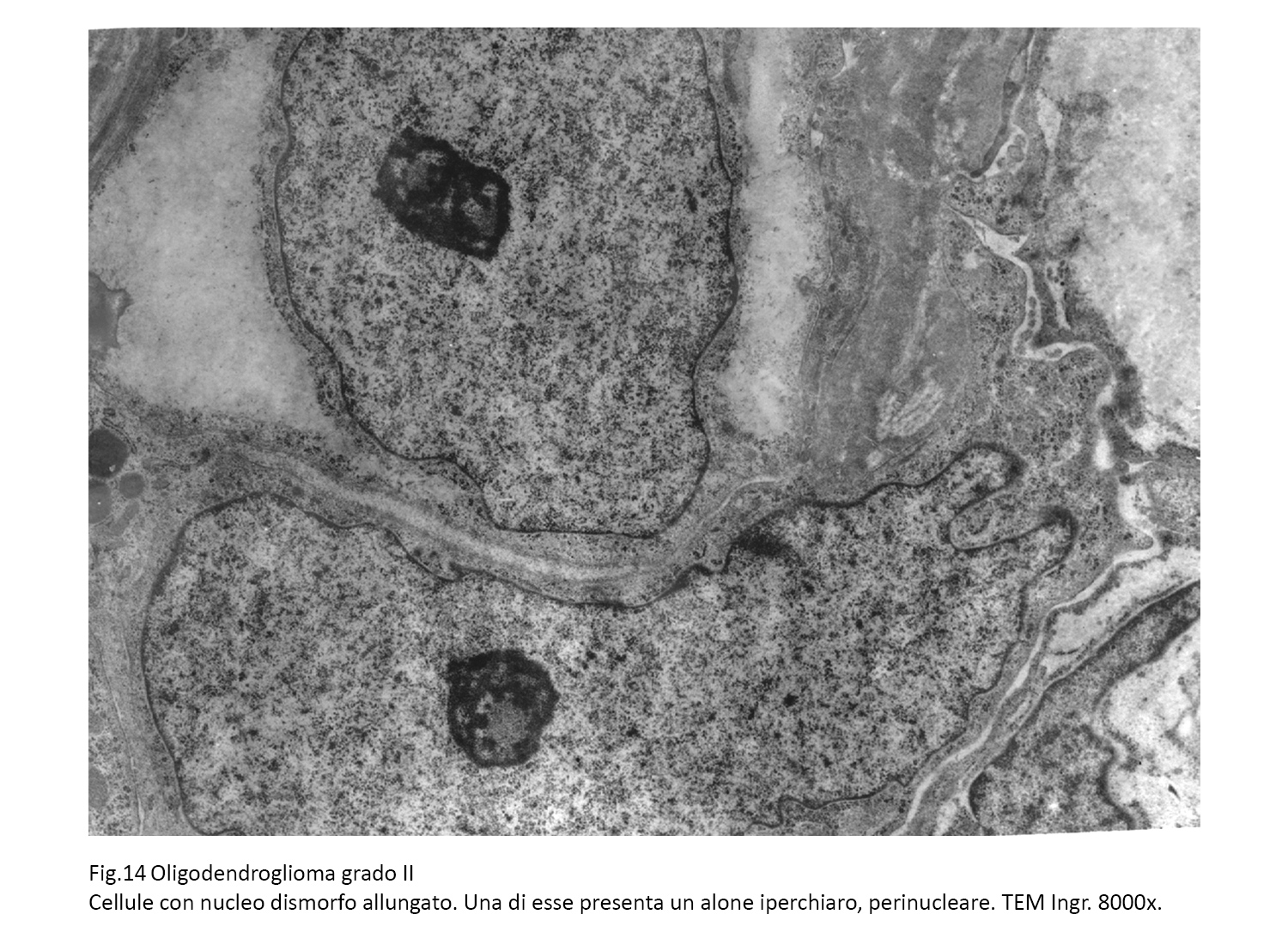
Le cellule dell’oligodendroglioma non rivelano espressività immunologiche per le GFAP; se si repertano cellule con citoplasma immunoespressivo per tale proteina, sono da considerare elementi astrocitari reattivi o intrappolati nella neoplasia. Sono state osservate positività immunologiche per la GFAP in casi di oligodendrogliomi glio-fibrillari oppure oligodendrogliomi minigenistocitici, si ipotizza che questi elementi siano forme transizionali o intermedie tra oligodendrociti (W.H.O., 2007).
Fig.15 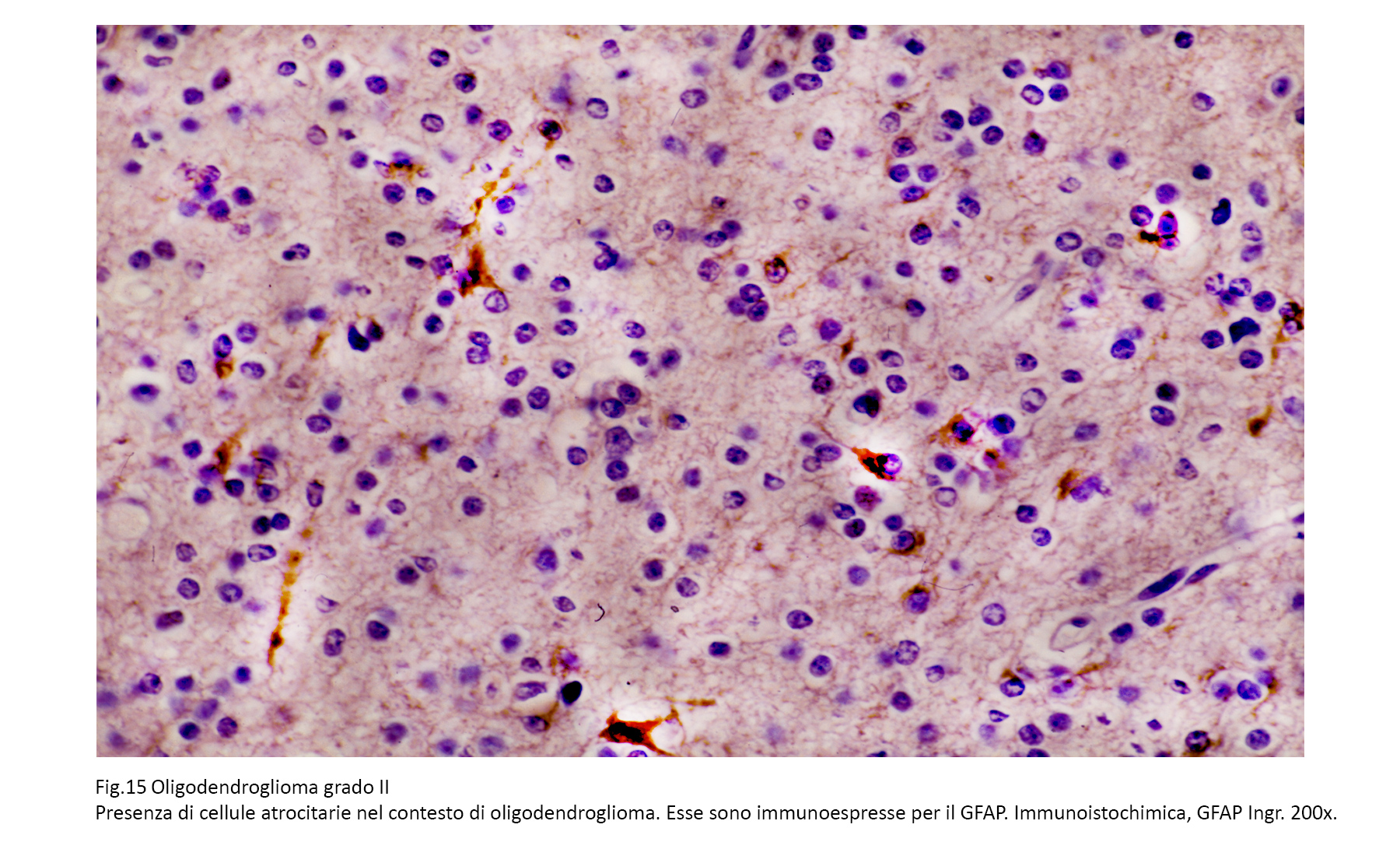
Le indagini genetico-molecolari nei casi di oligodendrogliomi sono importanti sia per la definizione diagnostica sia per valutare in prospettiva la prognosi e per controllare la efficacia della terapia. Dalla letteratura sono riportati i seguenti parametri genetico-molecolari per la definizione degli oligodendrogliomi grado II.
Delezione del braccio corto del cromosoma 1 (1p)
Delezione del braccio lungo del cromosoma 19 (19q)
Mutazione dei geni CIC e IDH.1
Polisomia del cromosoma 7
Perdita dei cromosomi 9, 10.
Amplificazione del gene EGFR
MMp1 e MMp9 positività
Il gene MGMT è in correlazione alla codelezione di 1p/19q
(Am. J. Path., 2007, 179, 1638-44)
(Neuro Oncol. 2009, 11, 737-48)
(Neuro Oncol. 2013, 15, 1244-1250)
(Neuro Oncol. 2014, 16, 1230-39)
(J. Neuro Oncol. 2014, 120, 131-41)
E’ necessario tener conto che esistono interazioni funzionali tra geni; infatti, è documentata una mutazione del gene CIC e FUBP1 con una mutazione di IDH1/2 associata alla codelezione 1p199. (J. Pathol. 2012, 261, 7-14) (Curr. Opin. Oncol., 2012, 24, 687-93).
L’espressività del gene EGFR se è associata alla codelezione 1p/19q acquisisce valori più alti ed esprime una prognosi sfaroreviole. (Am. J. Path. 2011, 179, 1631-44).
I membri MMP-2 e MMP-9 della famiglia della metalloproteinosi svolgono un ruolo importante negli oligodendrogliomi a favorire lo sviluppo della neoangiogenesi, quali uno dei meccanismi di sviluppo, infiltrazione e diffusione del tumore. (Anticancer Res. 2003, 23, 3937-44).
Il profilo genetico-molecolare di pazienti in età pediatrica colpiti da oligodendrogliomi si rivela una ridotta significanza.
Infatti nella prima decade di vita, la codelezione 1p/19q è assente ed è un risultato raro nell’età infantile. Allo stesso modo risultano non accertabili le mutazioni MGMT e IDHI. (Neuro-Oncol. 2011, 13, 1099-1106).
Gli oligodendrogliomi recidivati presentano caratteri morfologici, immunoistochimici e genetico-molecolari diversificati rispetto alle forme primitive. Nei casi recidivati la popolazione cellulare è iperdensa, ha una disposizione spaziale diffusa, le cellule sono pleomorfe e si rileva un numero maggiore di mitosi tipiche ed atipiche. Ciò è associato a una immunoespressività elevata per il Ki67 e per la p53. Infine il profilo genetico-molecolare evidenzia i seguenti caratteri:
Perdita dell’eterorigosità 1p/19q
Mutazione dei geni IDH1/2
Mutazione dei geni CIC e FUBP1
(Neuro Oncol. 2011, 13, 1099-1106)
(Brain Tumor Pathol., 2013, 30, 151-9)
(Acta Neuropathol., 2012, 123, 853-60)
Am. J. Surp. Pathol., 2014, 38, 1058-70)
Sono descritti in letteratura casi rari di oligodendrogliomi associati a neurocitomi e/o a gangliocitomi.
La diagnosi di questi casi casi richiede una procedura coordinata ed integrata di istopatologia, di immunoistochimica e di genetica molecolare.
L’istopatologia serve a evidenziare le diversità morfologiche dei citotipi presenti nella neoplasia. L’immunoistochimica evidenzia espressività specifiche per gli oligodendrociti mediante l’Oligo ½ e le espressività immunologiche caratterizzanti i neurociti o i gangliociti mediante la sinaptofisina, la Neun, La genetica molecolare completa la linea delle indagini diagnostiche mediante gli accertamenti riguardanti la codelezione 1p/19q, le mutazioni del gene YDH/1, la deidrogenasi 1.
(Brain Tumor Pathol. 2012, 29, 221-8)
(Human Pathol. 2013, 44, 2353-9)
L’oligodendroglioma anaplastico (oligodendroglioma grado III) può insorgere primitivamente come tale; oppure, ed è l’evenienza più frequente, può derivare da un pre-esistente oligodendroglioma di II grado.
Esso, sviluppandosi rapidamente con accrescimento infiltrativo provoca dissociazioni, necrosi e si sostituisce progressivamente alla neoplasia pre-esistente.
I margini sono molto incerti e sfumati e la neoplasia è sede di focolai emorragici, di aree necrotiche, di foci calcifici e di microescavazioni. Al microscopio esso appare quale neoplasia ad alta densità cellulare e priva di un’architettura per una disposizione diffusa degli elementi. Questi sono tra loro a mutuo contatto senza alcun segno morfologico di coesi. Le cellule sono pleomorfe per il loro profilo fusato, irregolare, poligonale, con aspetti anche simil-sarcomatosici e per la presenza di cellule giganti plurinucleate.
La quota citoplasmatica è espressa invece i nuclei sono voluminosi, compatti, nucleolati, irregolari, atipici e frequentemente sono in attività mitotica tipica ed atipica documentabile mediante una elevata espressività di Ki67.
A questa proliferazione cellulare si accompagna un processo di neoangiogenesi avente una distribuzione irregolare, asimmetrica e una struttura vascolare semplificata.
L’EORTC (European Organization for researrch and treatment of cancer) indica che i dati genetico molecolari diano maggiori certezze rispetto a quelli istopatologici per la diagnosi e la istogenesi degli oligodendrogliomi anaplastici.
Questi dati genetico-molecolari sono così definiti
Codelezione di 1p/19q
Polisoma del cromosoma 7
Perdita del cromosoma 10
Delezione del braccio corto cromosoma 9
Amplificazione del gene EGFR
(Neuro-Oncol., 2009, 11, 737-46)
A questi accertamenti sono state aggiunte le seguenti mutazioni geniche.
Mutazione del gene CDKN2C
Mutazione dei geni CIC e FUBP1
Mutazione YDH1 e YDH2
Mutazione del gene PTEW
(Acta Neuropath. 2012, 123, 853-60)
(Am. J. Surg. Path, 2014, 38, 1058-70)
(Neuro Oncol., 2013, 15, 775-782)
Secondo l’EORTC si ritrovano due sottotipi di oligodendroglioma anaplastico:
- Il primo tipo ha la perdita di 1p/19q; è localizzato a livello del lobo frontale ed è a prognosi favorevole.
- Il secondo tipo ha anche amplificazione di EGFR, alterazioni del cromosoma 7 e/o 10, ha localizzazione extra-frontale, ha un profilo genotipico oligo-astrocitario ed è a prognosi meno favorevole. (Neuro Oncol. 2009, 11, 737-46).
- Frequentemente si pongono quesiti di diagnosi differenziale tra oligodendrogliomi anaplastici e GBM a piccole cellule. In questi casi è necessario avvalersi dopo i reperti di istopatologia e di immunoistochimica, di dati genetico-molecolari con particolare riferimento alle mutazioni YDH1/2, alla alterazione PTEN e alle codelezioni 1p/19q. (Brain Tumor Path. 2014, 31, 118-23).
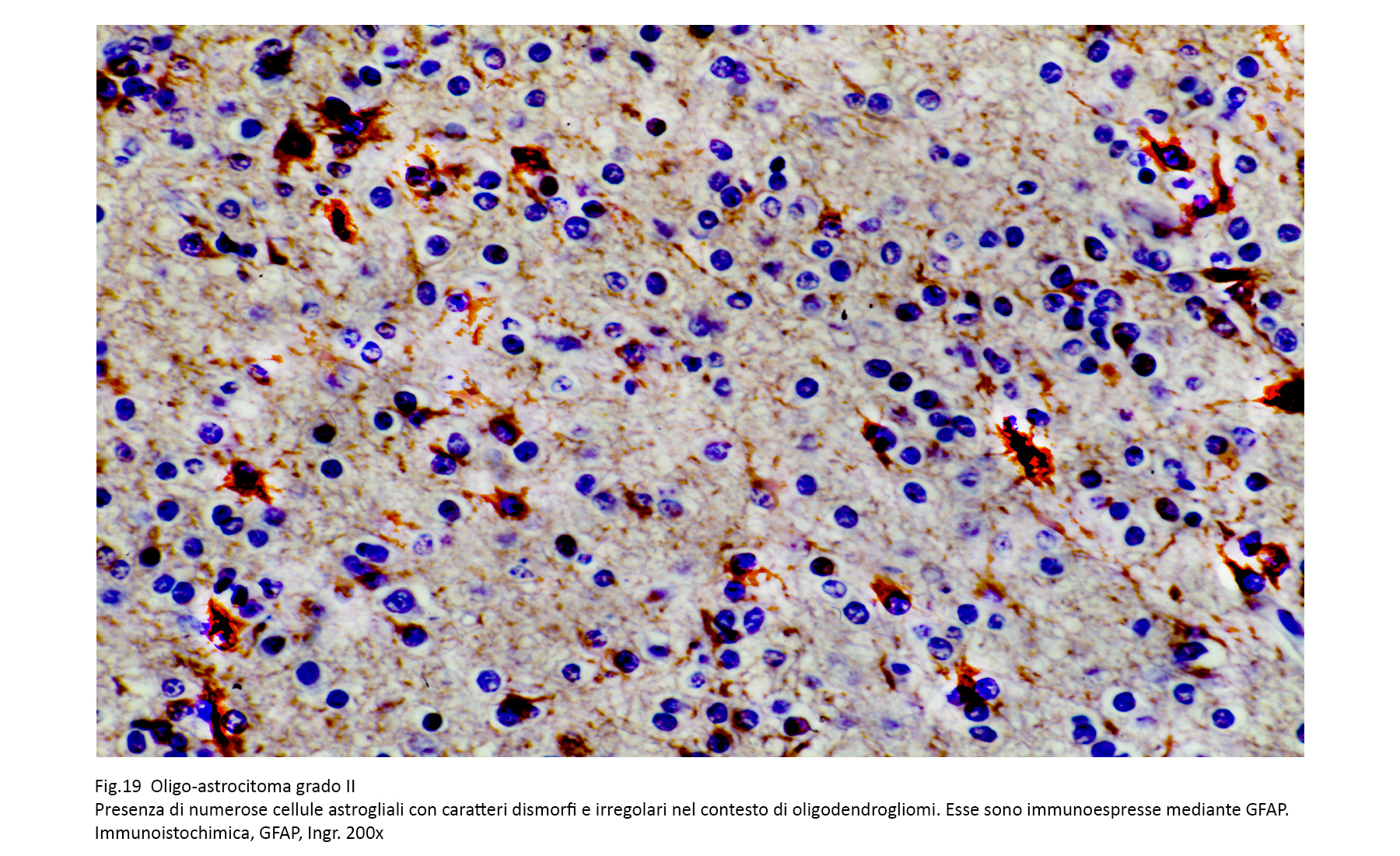
L’Oligoastrocitoma è una neoplasia costituita da due istotipi, morfologicamente differenziabili, dei quali uno ha i caratteri dell’oligodendroglioma e l’altro quello dell’astrocitoma. Sul piano architettonico questa neoplasia si appalesa secondo due varianti: la prima è data dalla coesistenza spazialmente separabile di due aree neoplastiche, una oligodendrogliale, l’altra astrocitaria; la seconda è formata da una stretta commistione, non separabile, di cellule oligodendrogliali ed astrocitarie.
Nell’ambito di questa popolazione mista possono ritrovarsi anche gruppi cellulari con caratteri genotipici intermedi oligo-astrocitari.
La componente astrocitaria può essere differenziata nei vari sottotipi (fibrillare, protoplasmatica, genestocitico, etc.) e la quota oligondrogliale può essere di II o III grado.
Sono riportati in letteratura casi di oligo-astrocitomi anaplastici; questi istotipi evidenziano una struttura composita (oligodendrogliale e astrocitaria) avente i caratteri morfobiologici di malignità quali ipercellularità, pleomorfismo, atipie nucleari, alto indice mitotico, focolai necrotici, neoangiogenesi.
Questa neoplasia mostra caratteri eterogenei perché oltre ai citotipi dianzi ricordati si possono repertare elementi con caratteri genotipici intermedi per incompleto livello di differenziazione.
Pertanto sono indispensabili indagini di imunoistochimica e di genetica molecolare per addivenire a una diagnosi di certezza.
Mediante la immunoistochimica è possibile identificare la quota astrocitaria attraverso la espressività immunologica per la proteina GFAP.
Fig.16 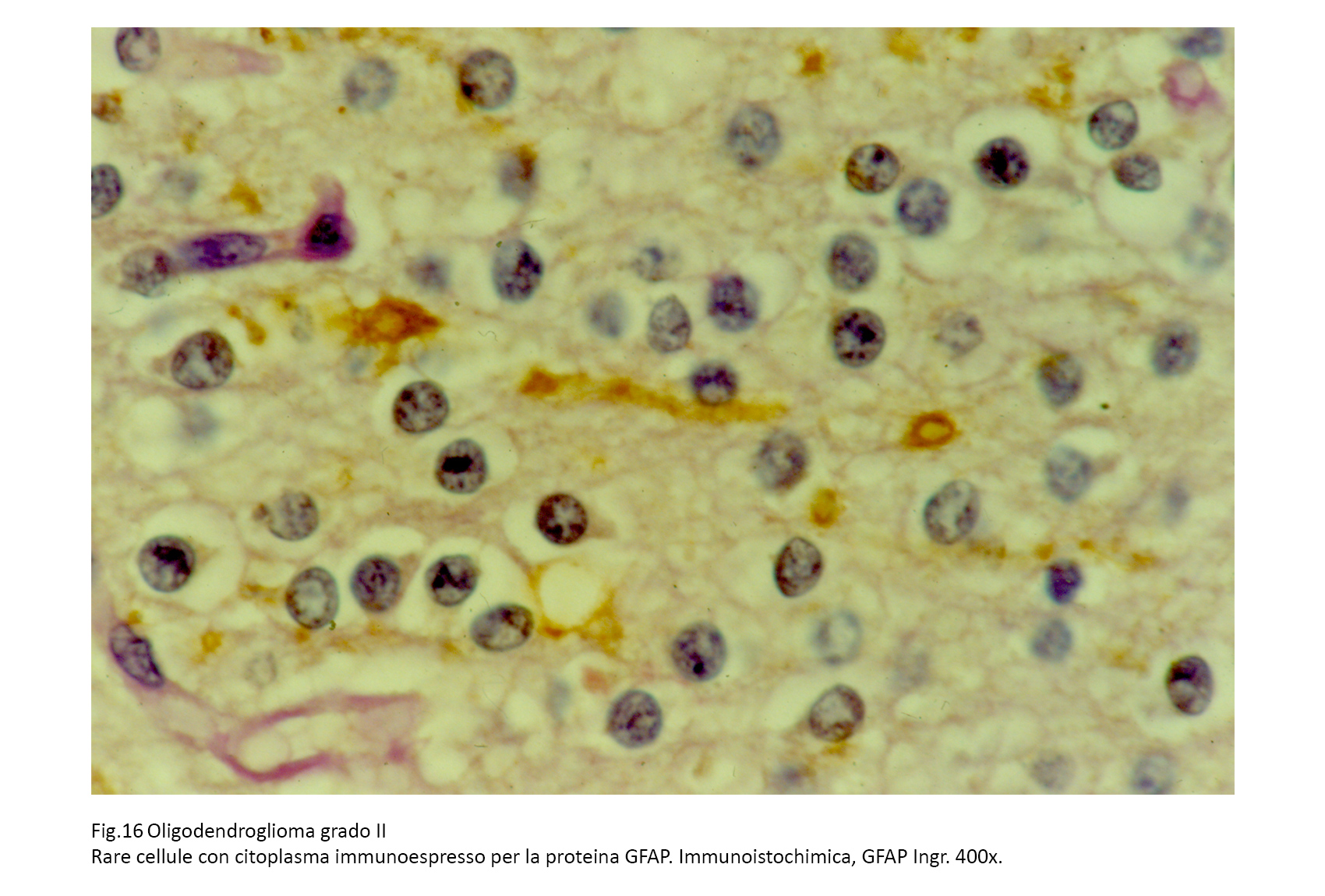
Inoltre si può procedere alla delimitazione della componente oligodendrocitica mediante le specifiche espressività immunologiche di tali cellule. Infine si può stabilire l’indice di attività proliferativa attraverso la valutazione della positività nucleare del Ki67. Le indagini genetico-molecolari evidenziano i seguenti caratteri utili per la definizione della componente oligodendrogliale.
Perdita di 1p/19q Perdita dei cromosomi 10 e 11p
Mutazione di TP 53 Amplificazione del gene EGFR
Perdita di 9p Acta Neuropathol.2012, 123, 853-60
(Neuro Oncol. 2013, 15, 775-82)
Su la base dei dati genetico-molecolari gli oligoastrocitomi anaplastici sono distinti in due sottotipi:
Il primo è caratterizzato soprattutto dalla perdita di 1p/19q e pertanto si riconosce una dominanza di cellule oligodendrogliali.
Il secondo ha come dato di maggiore rilievo la perdita di 17p e la mutazione di TP53, quali espressione genetica di una dominanza di astrociti.
(Am. J. Pathol. 2010, 177, 2708-14).
In letteratura vengono riportati casi rari di oligo-sarcoma. Questa neoplasia, apparentemente bifasica è costituita da elementi oligodendrogliali e da cellule simil-mesenchimali. Sono descritte di questo tumore due varianti:
la prima è formata da due componenti tra loro ben distinguibili e separabili, la seconda svela una disposizione strutturale strettamente commista in modo disordinato. E’ stato accertato che reperti di oligo-sarcomi possono subentrare a casi di oligodendroglioni anaplastici recidivati.
La quota simil-mesenchimale appare a livello istopatologico quasi sempre formata da cellule fusate atipiche, avente caratteri fibrosarcomatosici. Questa quota rivela alle indagini di immunoistochimica espressività per la Vimentina, per l’EMA, per l’actina muscolo liscio, per la proteina S-100.
Infine, le ricerche di genetico-molecole hanno svelato che le suddette due componenti, fenotipicamente differenti hanno lo stesso profilo (co-delezione di sp/19q, perdita del gene JDH1, etc.) genetico, confermando così la loro monoclonalità.
(Clin. Neuropathol. 2013, 32, 165-70)
(Pathol. Res. Pract. 2012, 208, 750-5)
Fig.17 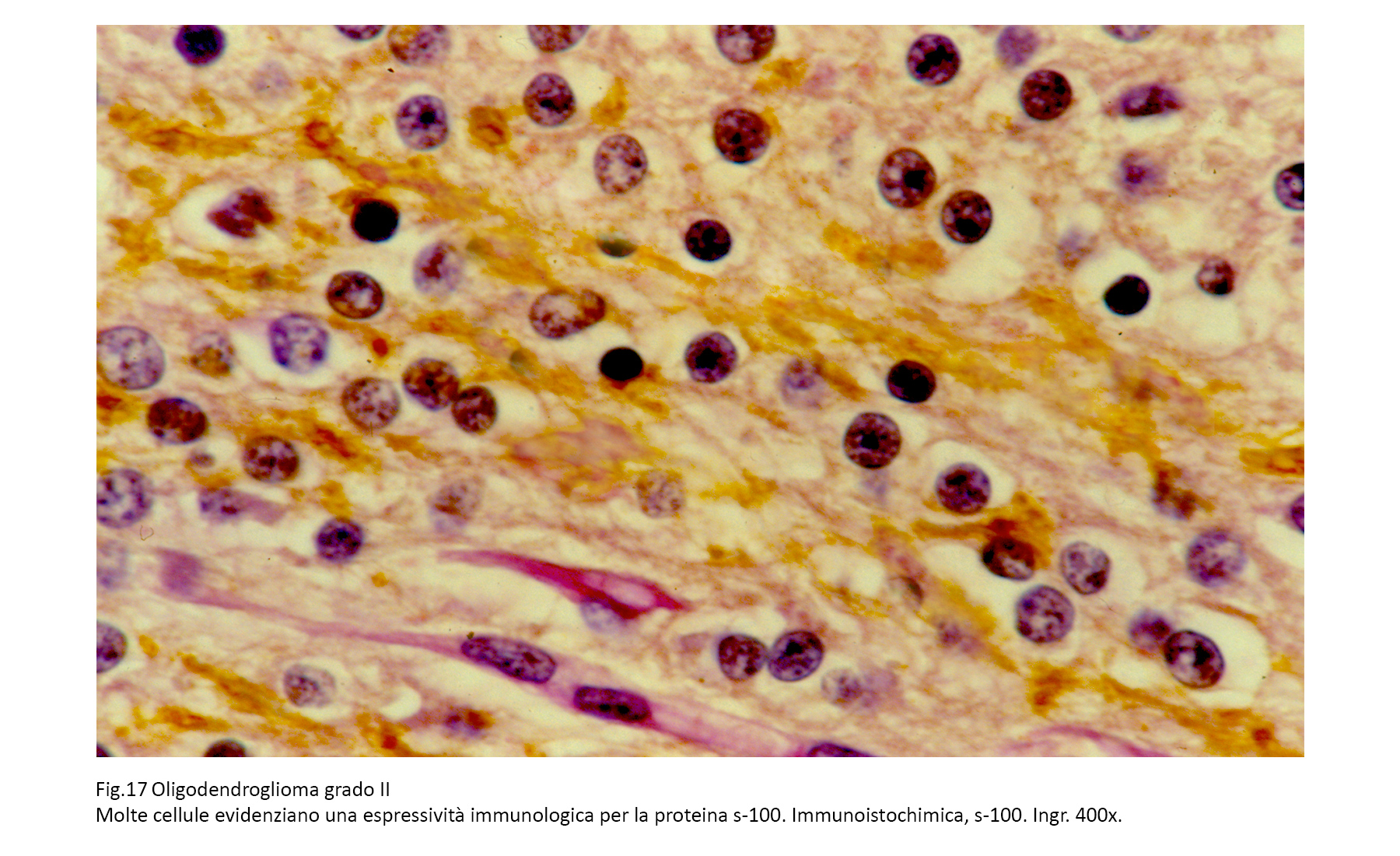 Fig.18
Fig.18 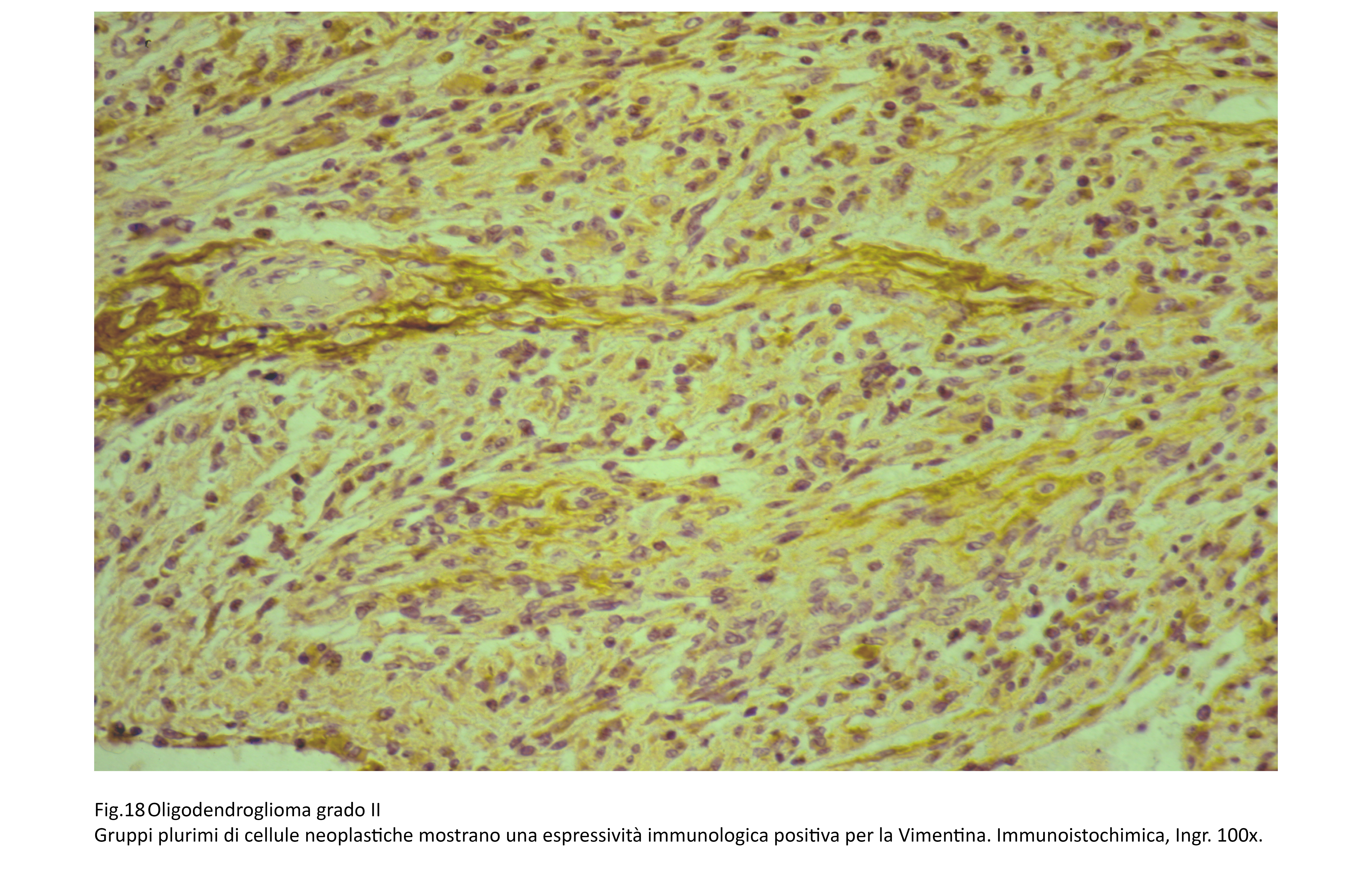 Fig.19
Fig.19