Il glioblastoma multiforme (GBM) è un tumore ad alta malignità, di natura astrogliale a crescita infiltrativa; esso è caratterizzato da alta attività mitotica, da notevole pleomorfismo cellulare, da marcata neoangiogenesi, dalla esistenza di macronecrosi e da micronecrosi con contorno di cellule a pseudo-palizzata.
CARATTERI MACROSCOPICI
Il glioblastoma multiforme si forma e si sviluppa, nella maggior parte dei casi, a livello della sostanza bianca degli emisferi cerebrali. I lobi frontali sono coinvolti con una incidenza maggiore, seguono per frequenza i lobi temporali, quindi quelli occipitali; è stata riportata in letteratura una localizzazione non numerosa a livello del cervelletto e un raro riscontro a carico del midollo spinale e dei nuclei della base.
Il focolaio glioblastomatoso inizia abitualmente nel contesto della sostanza bianca, ma può formarsi anche in sede sottocorticale con conseguente precoce slargamento, deformazione,e ispessimento delle circonvoluzioni.Il GBM si accresce e si sviluppa secondo le modalità di tipo infiltrativo, interessa i tessuti e le strutture contigue, si riversa nel torrente circolatorio ematico, si apre nella via liquorale, disegnando quadri anatomo-clinici diversi a seconda delle vie di penetrazione e di diffusione.Si repertano, con frequenza, casi in cui il GBM invade il corpo calloso per poi diffondersi e svilupparsi a livello dell’emisfero controlaterale (aspetto a farfalla); può infiltrare a tutto spessore la corteccia cerebrale e occupare gli spazi sub-aracnoidei, permeare le meningi molli e stratificarsi su la faccia interna della dura; non è esclusa la evenienza che possa superare la barriera ependimale, e invadere i ventricoli, ostruendoli.Il GBM può appalesarsi come una neoformazione isolata oppure come focolai plurimi disseminati nel contesto del sistema nervo centrale. Questa seconda modalità può essere conseguente alla esistenza di focolai neoplastici plurimi autonomi ed indipendenti (GBM multicentrico); oppure allo sviluppo di focolai neoplastici plurimi correlati e collegati biologicamente con un focolaio di partenza (GBM multifocale).
In letteratura sono descritti su la base di dati anamnestico-clinici correlati da reperti anatomo-patologici, GBM primitivi e GBM secondari; i primi sono quei casi insorgenti come tali fin dall’inizio, i secondi sono quei reperti che si formano e si sviluppano su un presistente astrocitoma di II o di III grado.
Il GBM ha dimensioni variabili in rapporto alla epoca di osservazione; nel suo accrescersi può occupare l’intero emisfero interessato dal tumore. La forma è irregolare perché lo sviluppo si realizza seguendo i poli di accrescimento ove sono maggiormente concentrate le cellule in attività mitotico- proliferativa. I margini sono sfumati e indistinti per le modalità di crescita del GBM; a tale proposito si riconosce la esistenza di un margine determinabile all’esame macroscopico il quale appare più circoscritto rispetto a quello rilevabile al microscopio, indicante le reali dimensioni del tumore.
La massa neoplastica ha un aspetto variegato per colore e consistenza con notevoli differenze da zona a zona nell’ambito dello stesso campione; al taglio si nota abitualmente che la fascia periferica è compatta ed è di colore grigio-biancastro con punteggiature emorragiche invece l’area centrale è poltacea e ha un colore rosso-giallastro con escavazioni per processi necrotico-emorragici.Il quadro macroscopico di base può modificarsi per la comparsa di processi secondari quali i focolai flogistici, la neo-angiogenesi, l’edema, gli stravasi emorragici, la reazione microgliale,e la proliferazione di componenti mesenchimali di diverso profilo.
Il GBM è stato considerato da sempre una entità nosografica unitaria sebbene fossero state accertate e descritte notevoli variabilità a livello morfologico, clinico e terapeutico.La citomorfologia dei GBM è molto diversificata ed è possibile repertare cellule anaplastiche, cellule di piccola taglia, cellule giganti uni o multinucleate, cellule gemistocitiche, cellule bipolari, cellule fusate, cellule stellate, cellule epiteliomorfe, cellule sarcomatosiche, cellule metaplasiche.Queste variabilità non sono state considerate tali da mettere in dubbio la identità formale di questa neoplasia; si è proceduto al riconoscimento della esistenza accanto all’istotipo-base anche di diversi sottotipi e di alcune varianti.
Questa interpretazione unitaria del GBM è stata profondamente rivisitata e modificata dopo le recenti acquisizioni molecolari- genetiche. I dati ottenuti da queste ricerche hanno consentito di affermare che il GBM non sia da considerare una entità unitaria bensì un gruppo di neoplasie tro loro ben distinte.
Queste ricerche molecolari e genetiche hanno documentato nei GBM livelli diversi di espressività genetiche, anomalie cromosomiche, mutazioni geniche, instabilità genetiche, situazioni tutte che si riflettono su la morfologia, la biologia, il quadro clinico e la responsività dei singoli casi di GBM (Nature, 455, 1061-68, 2008).
A convalidare questa nuova situazione diagnostico-dottrinale è utile riportare come esempio lo stato morfologico-molecolare dei GBM primitivi e dei GBM secondari. Queste neoplasie sono indistinguibili a livello istopatologico,ma risultano essere due entità diverse secondo i reperti molecolari.
Infatti il profilo molecolare dei GBM primitivi e dei GBM secondari si presenta a un esame analitico così diversificato:
GBM primitivo:
Amplificazione di EGFR, CDK4, MDM2, MDM4
Mutazione: PTEN, TP53
Monosomia:10
Delezione: omozigote CDKN24
Iperespressività: NARGE
GB secondario:
Iperespressività di PDGFRA, EMP3, MGMT
Mutazione: TP53, IDH1, PTEN
Amplificazione: EGFR, MDM2
Ipermetilazione del gene: RBI
Perdita dell’allele: 199 e 139
(American Journal of Clinical Pathology, 131, 25763, 2009),(Korean J. Pathol. 47, 541-48, 2013)
Allo stesso modo, la diagnosi differenziale tra GBM e astrocitoma anaplastico trova una più sicura risoluzione se oltre la valutazione istopatologica si aggiunge quella molecolare.I criteri istopatologici permettono di distinguere i glioblastomi multiformi dagli astrocitomi anaplastici per la presenza di focolai di necrosi, per la proliferazione dei vasi e per la estrema eterogeneità delle popolazioni cellulari.Questi elementi di diagnosi differenziale sono attualmente integrati da osservazioni eseguite a livello molecolare; infatti è stato messa in evidenza una diversità del patrimonio molecolare dei GBM rispetto agli astrocitomi anaplastici. In particolare, è stato rilevato come dati specifici dei glioblastomi,rispetto agli astrocitomi anaplastici i seguenti reperti genetico-molecolari:
Iperespressività di CDKN3 e CHI3LI, VEGFR, FABP7.
Presenza di EMT
Iperregolazione di NAMPT, TOP2A, Laminina 8
Pub. on line , Jan 24,2014
The Cancer Genoma Atlas (TCGA) ha elaborato una classificazione su base molecolare dei GBM nella quale sono inserite nuove varianti o sottotipi considerati secondo la istopatologia gliomi di basso grado inglobanti, nel loro contesto gruppi di cellule riferibili a glioblastoma; questi gruppi cellulari sono svelati mediante metodiche genetico-molecolari e sono identificati quali aggregati che sviluppandosi manifestano a livello biologico-clinico una rapida evoluzione e a prognosi infausta.
Questa classificazione (TGCA) è l’espressione integrata di caratteri morfologici con dati genetico- molecolari e si articola nelle seguenti voci:
Glioblastoma classico
Gliosarcoma
Glioblastoma a cellule giganti (gcGBM)
GBM fibrillare epiteliale
Astrocitoma a piccole cellule (SCA)
Glioblastoma con componente oligodendrogliale
Glioblastoma con primitivo carattere neuro-ectodermico (GBM-PNET)
Astrocitoma gemistocitico (GA)
Astrocitoma a cellule granulari (GCA)
Glioblastoma pediatrico
(Frontiers in Bioscience, 19, 1066-87.2014)
L’esposizione analitica dei vari sottotipi del GBM qui di seguito elaborata seguirà i criteri della suddetta classificazione,ritenuta al momento quella esauriente e completa. La denominazione di alcuni sottotipi (in conseguenza della integrazione tra reperti morfologici e molecolari) può provocare qualche disorientamento lessicale;al fine di superare questa difficoltà è stato ritenuto opportuno utilizzare per questi sottotipi una doppia terminologia: quella istopatologica (WHO) e quella molecolare (TGCA).
Glioblastoma classico (GBM)
Il GBM, definito classico, è caratterizzato dalla morfologia microscopica paradigmatica di riferimento poichè è fornito di tutte le micro-alterazioni e le micro-involuzioni considerate patognomiche di questo tumore. Pertanto, sarà dedicato a questa forma una descrizione analitica che servirà come linea-guida anche per gli altri sottotipi.
A piccolo ingrandimento il GBM classico appare ad alta densità cellulare ma privo di alcun disegno architettonico; esso è punteggiato da aree di necrosi, da focolai emorragici ed è sede di numerosi vasi distribuiti in modo irregolare e disordinato.
Fig.1 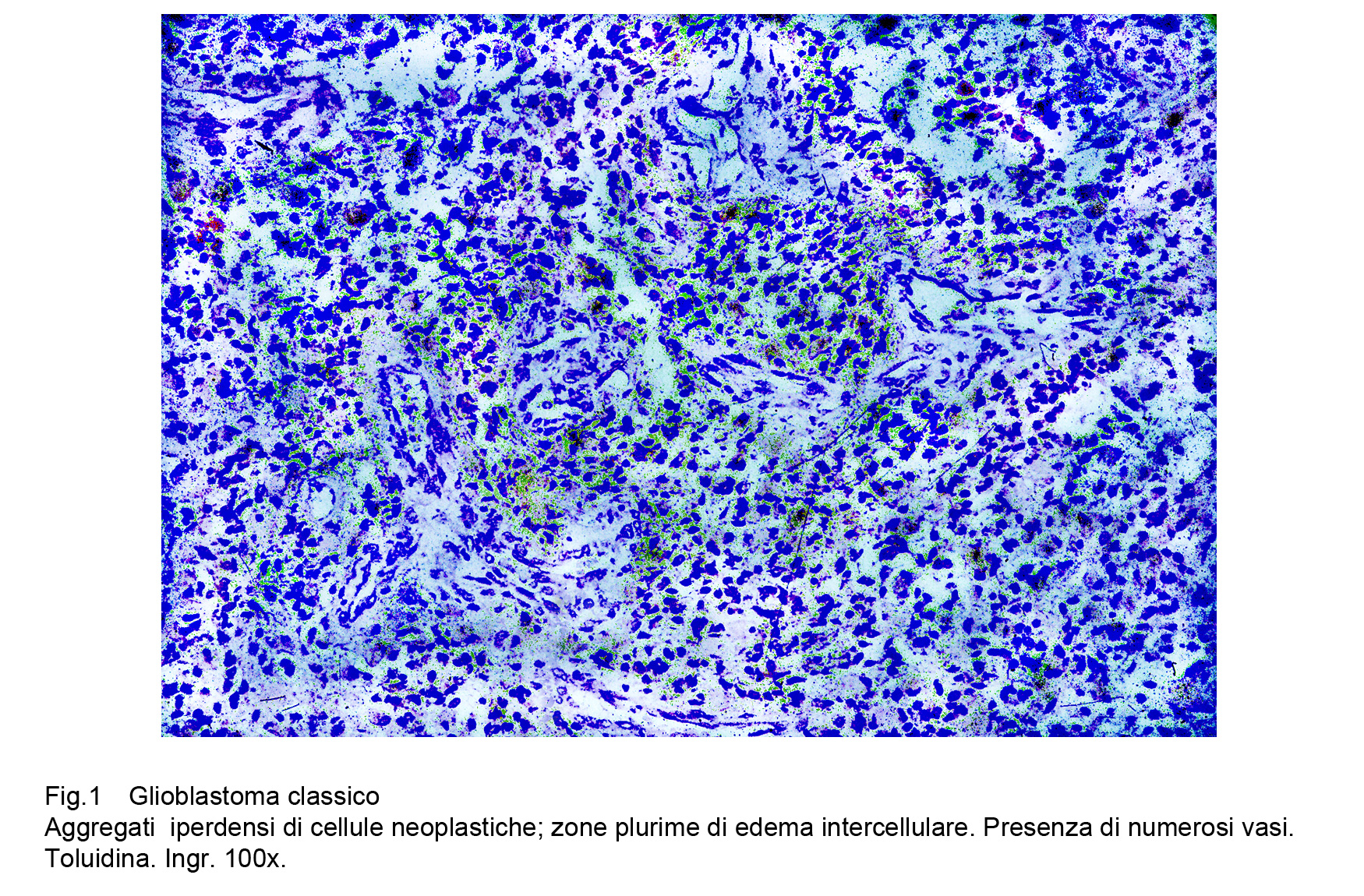
A maggiore ingrandimento si riscontra una popolazione cellulare pleomorfa per volumetria e morfologia; infatti in molti casi coesistono nello stesso campo microscopico cellule di piccola taglia assieme a cellule giganti uni-nucleate, bi-nucleate o mutlinucleate.La quota di citoplasma può essere sviluppata in diverso grado e i nuclei possono apparire voluminosi, vescicolosi, compatti, di forma bizzarra, ipercromatinici, mostruosi, con nucleolo prominente.Le cellule sono disposte tra loro a mutuo contatto in modo disordinato e caotico, ma sono prive di coesione.
Fig.2 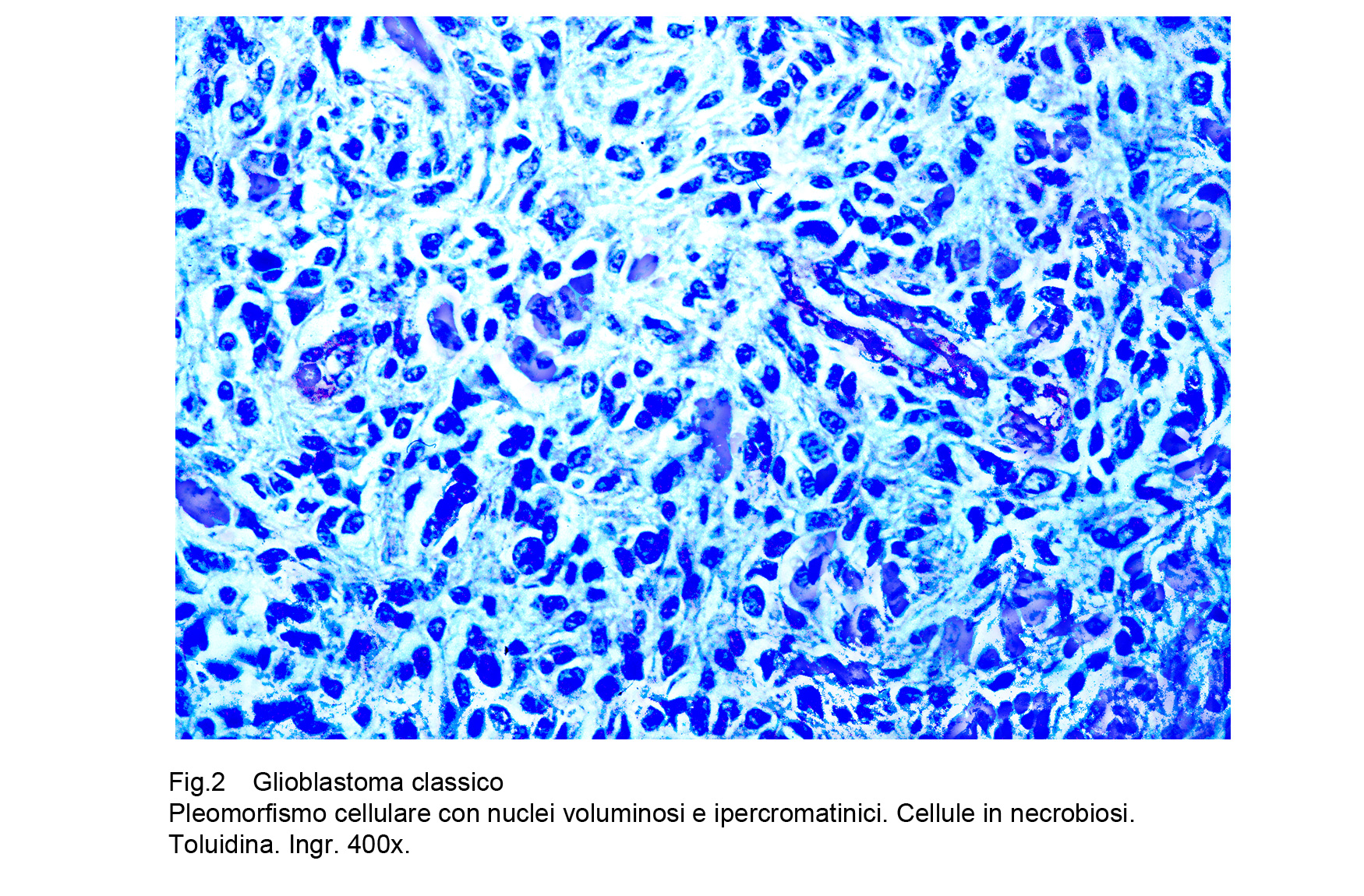 Fig.3
Fig.3 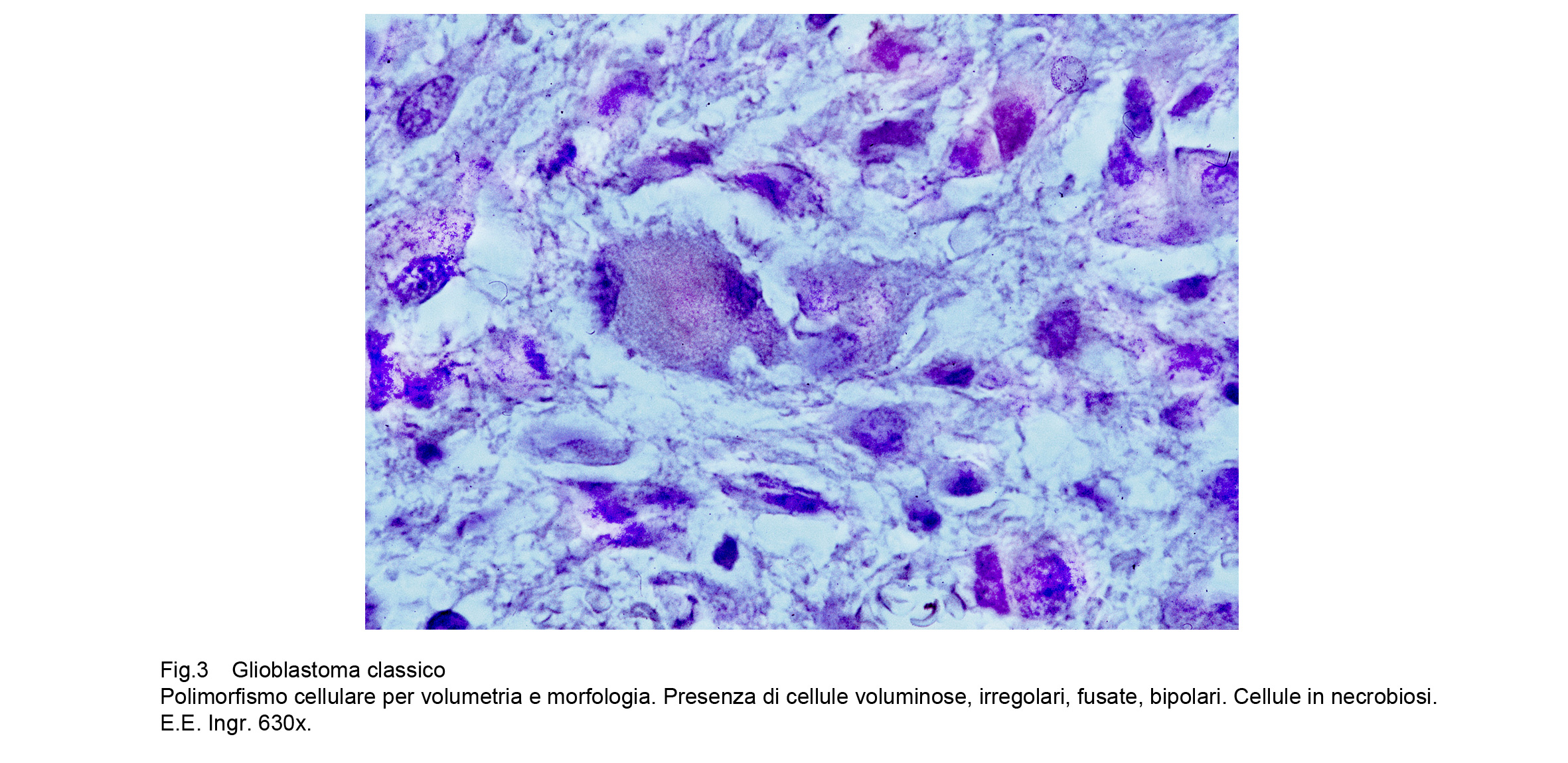 Fig.4
Fig.4 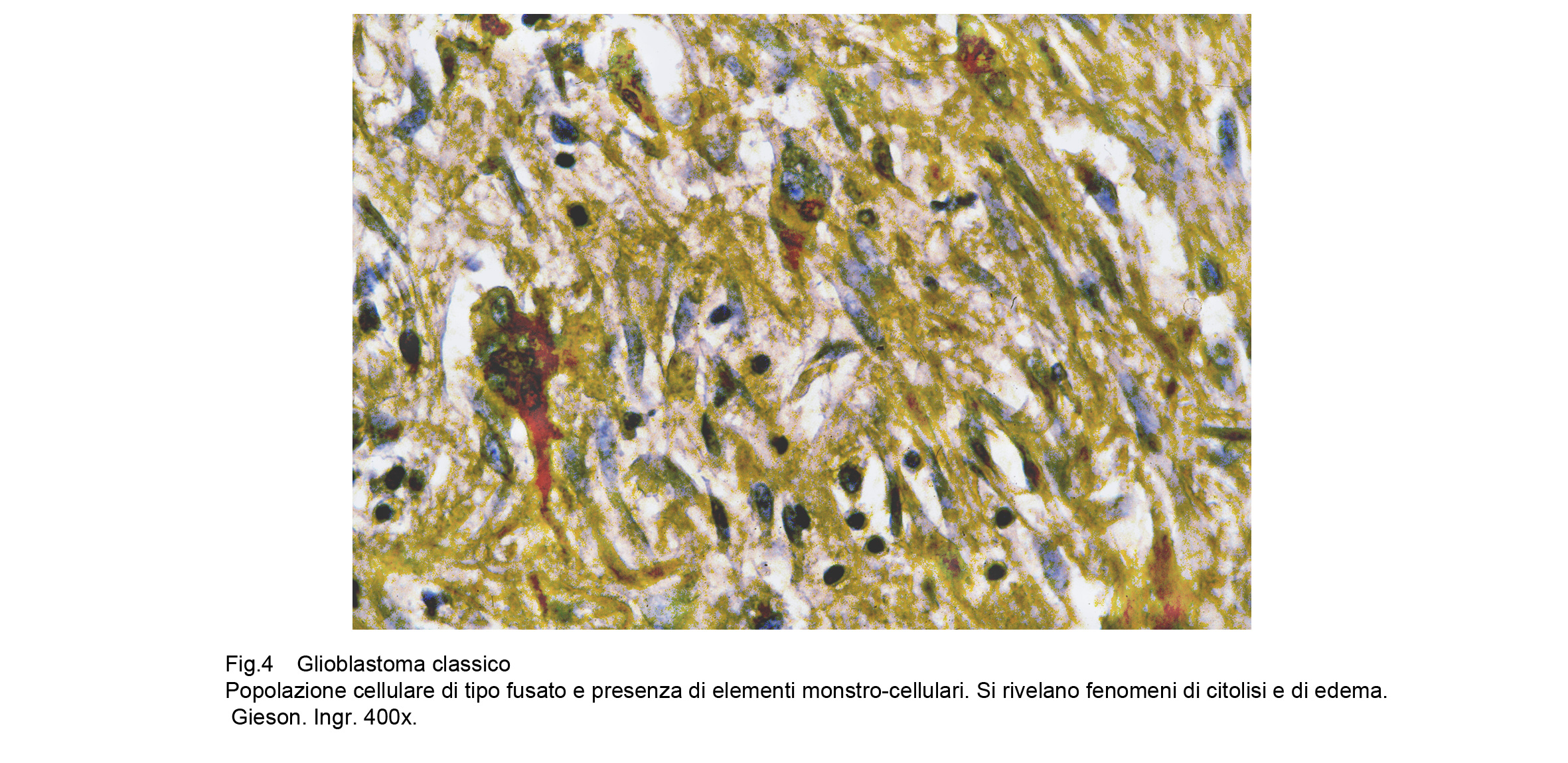
In tutto il GBM e soprattutto a livello delle aree di accrescimento si riscontra la presenza di numerose cellule in mitosi tipiche ed atipiche; commisti a tali elementi in attività proliferanti si repertano altri con nuclei in carioressi o con citoplasma vacuolizzato o ricolmo di gocce lipidiche o in necrobiosi.
Il GBM è sede di numerosi focolai di necrosi; questi sono distribuiti in modo apparentemente casuale e presentano due distinti aspetti morfologici.
Il primo tipo, è formato da focolai di macro-necrosi; questi sono estesi, hanno un profilo irregolare e sono privi di margini di demarcazione; essi sono sede di edema interstiziale e di stravasi emorragici e tendono ad ampliarsi secondo diverse direttrici. Se sono multipli e sono contigui (per il loro accrescimento centrifugo) possono confluire tra loro ,disegnando così vaste aree occupate da materiale amorfo, poltaceo, giallastro formato da lipidi e da detriti cellulari oppure dando così origine a escavazioni prive di pareti proprie.
Fig.5 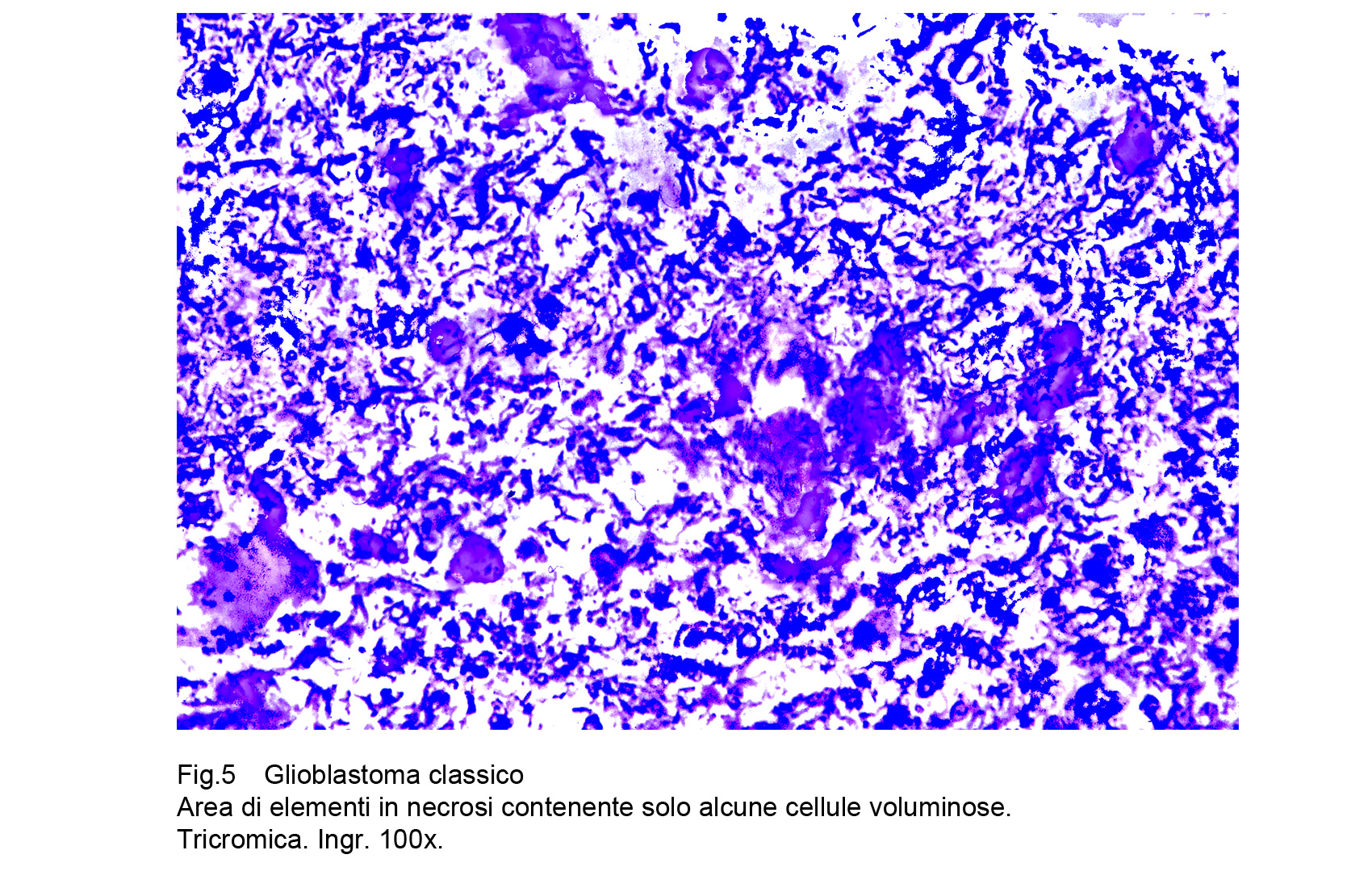
Il secondo tipo di focolaio di necrosi si manifesta quale area di piccole dimensioni avvolta da uno spesso cercine di cellule glioblastomatose disposte a strati sovrapposti, costruendo così un aspetto a pseudo-palizzata.
Fig.6 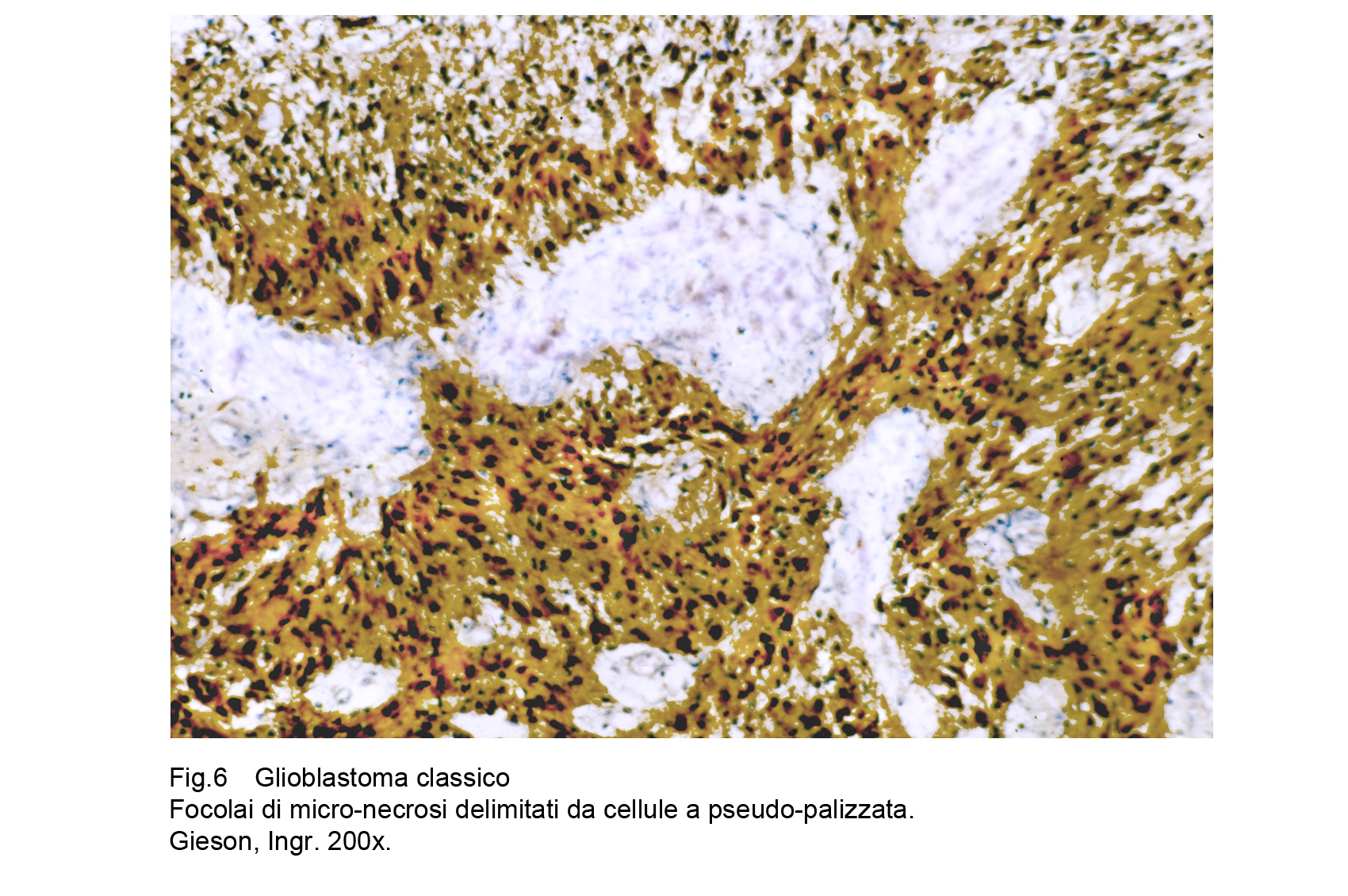
L’area necrotica assieme a materiale amorfo contiene ombre cellulari e sottili fibrille variamente disperse. Le cellule disposte alla periferia dell’area necrotica sono in attività proliferativa e a livello metabolico evidenziano un adattamento allo stato di ipossia che è stato indotto da preesistenti micro- -vasculopatie ostruttive che hanno causato la necrosi. Il riscontro di questi focolai viene considerato come reperto prognostico sfavorevole per la presenza di queste cellule glioblastomatose in attività proliferativa.
Il GBM è sede di una cospicua rete vascolare costituita da vasi di piccolo calibro e da capillari. Questa rete di neo-angiogenesi si sviluppa (assieme alla crescita tumorale) in modo disordinato, asimmetrico, formando anche aree pseudo-angiomatoidi o noduli sferoidali di aspetto glomeruloide.Questi vasi hanno pareti semplificate, sottili che sono in discordanza con il loro lume ampio, lacunare, tortuoso, ripieno di eritrociti e/o di trombi fibrinosi.A livello delle aree angiomatoidi questi aspetti sono molto accentuati con possibilità di lacerazioni e di spandimenti emorragici.Le microstrutture glomeruloidi sono gomitoli vascolari avvolti da fibrille reticolari o da cellule glioblastomatose.Particolare attenzione richiedono gli endoteli di questa componente vascolare; essi appaiono ipertrofici, alti, voluminosi e spesso per processi proliferativi si dispongono a strati sovrapposti.Al contrario dei vasi, i capillari sono ben strutturati , sono forniti di una membrana basale, di un rivestimento continuo di endoteli e sono avvolti da periciti.Frequentemente sono circondati anche da cuffie di elementi immunocompetenti o da anelli di cellule glioblastomatose che, superando la parete del capillare si riversano nel torrente circolatorio.Per subentranti processi involutivo-degenerativi le pareti dei vasi possono ispessirsi per fibrosi, per ialinosi, per microdepositi di sali di calcio. Questi processi fibro-ialonitici possono interessare anche le strutture peri-vascolari e possono associarsi a proliferazione di istiociti, e di macrofagi carichi di lipidi.
Fig.7 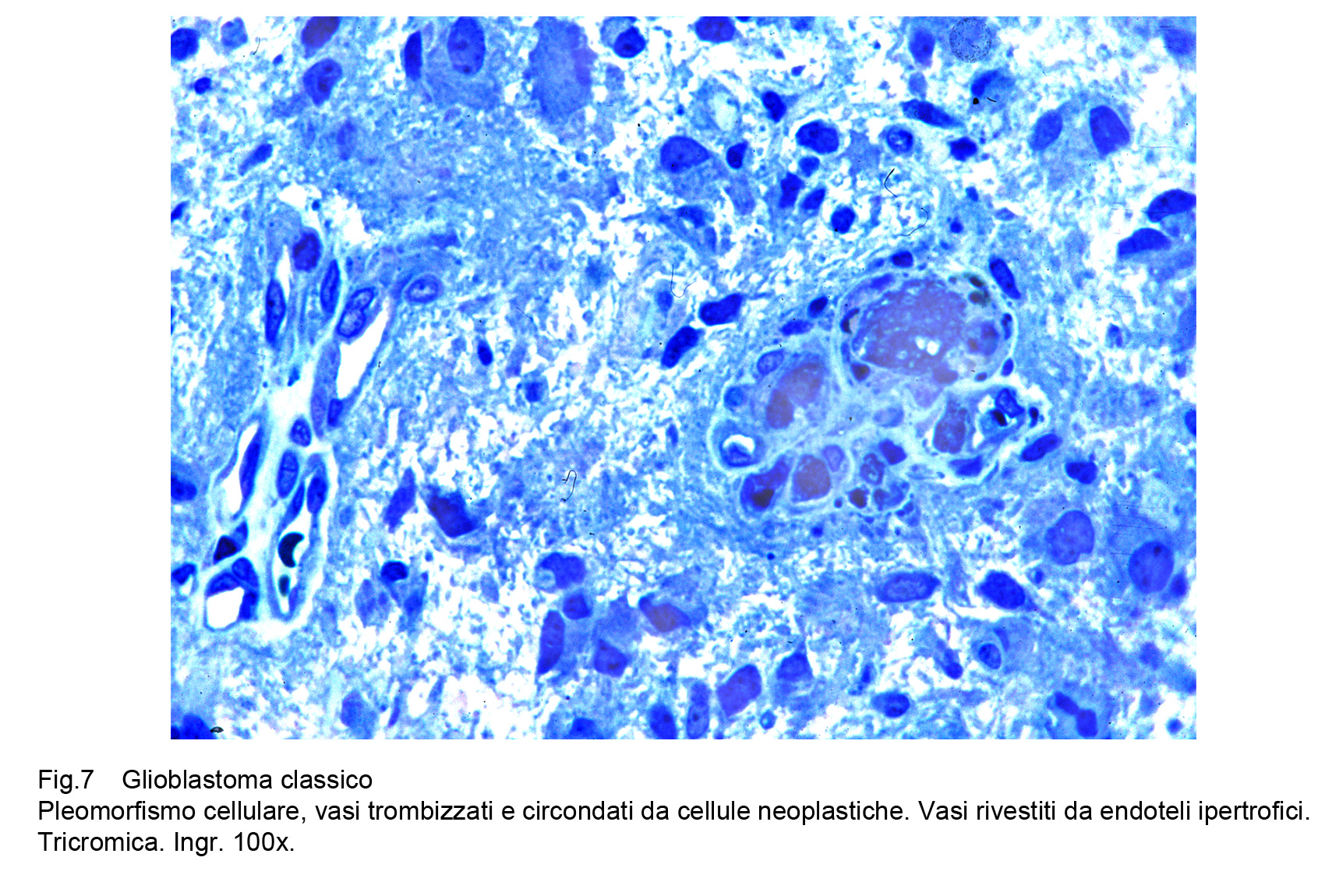 Fig.8
Fig.8 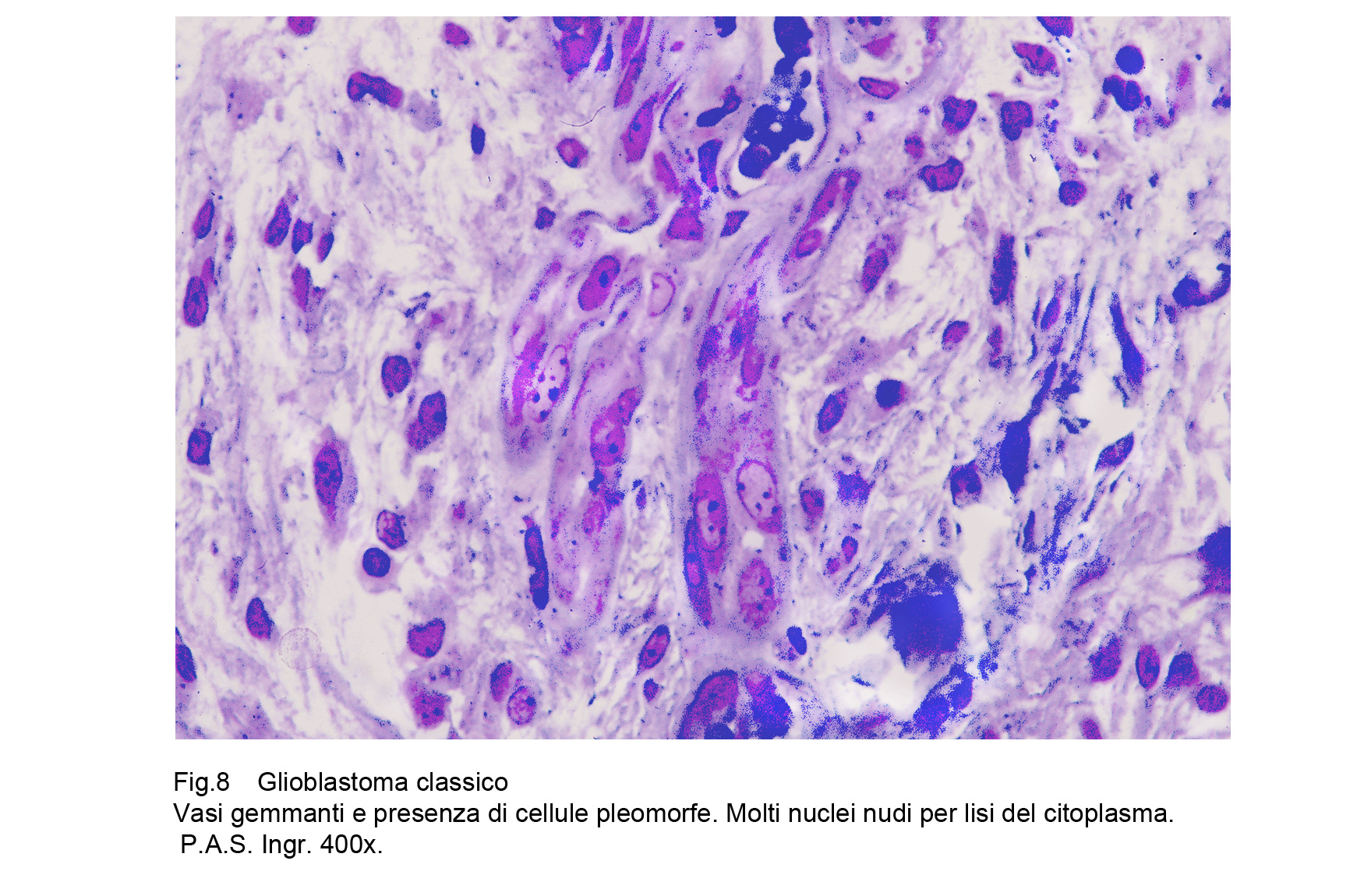 Fig.9
Fig.9 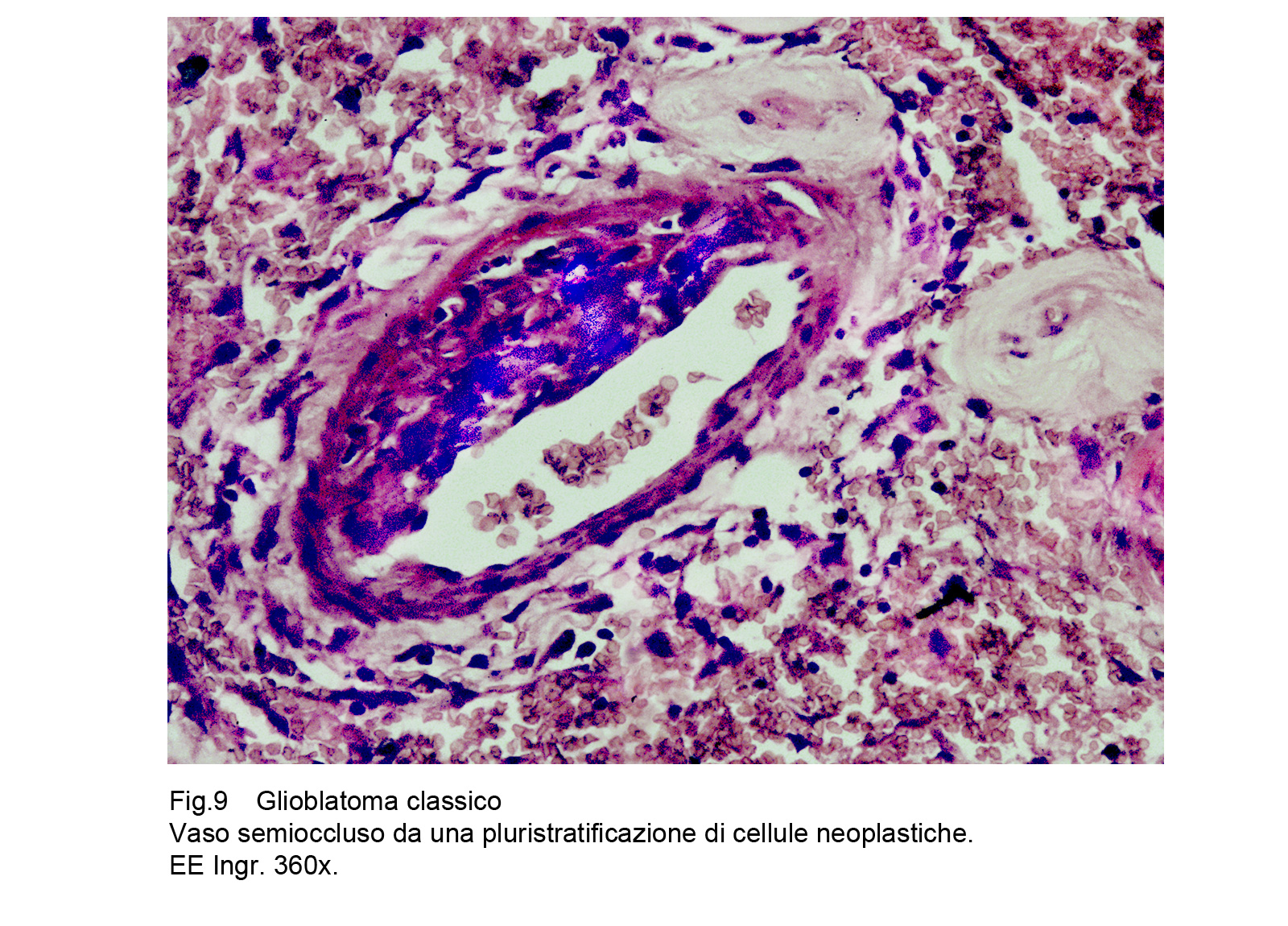
Fig.10 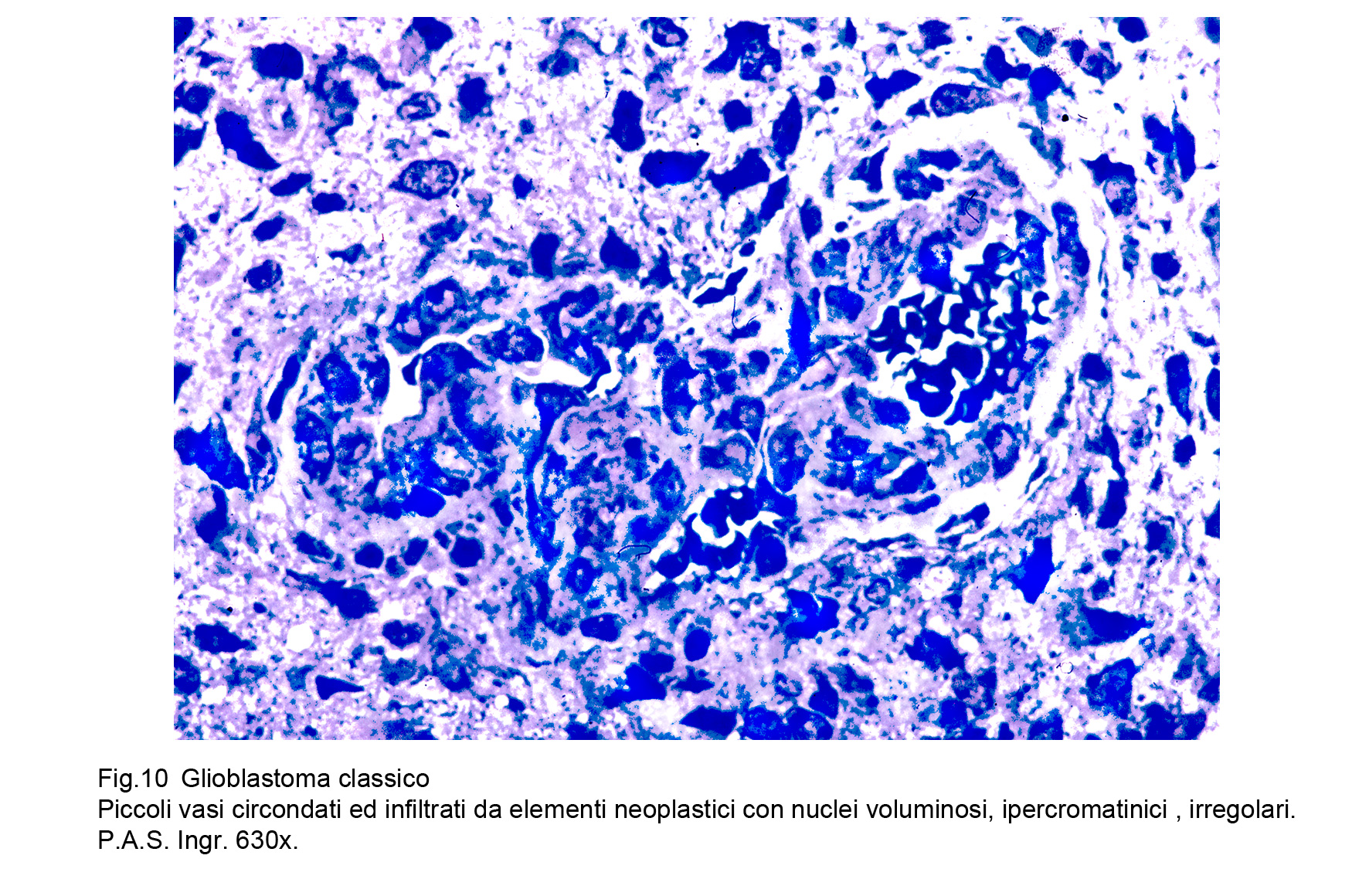 Fig.11
Fig.11 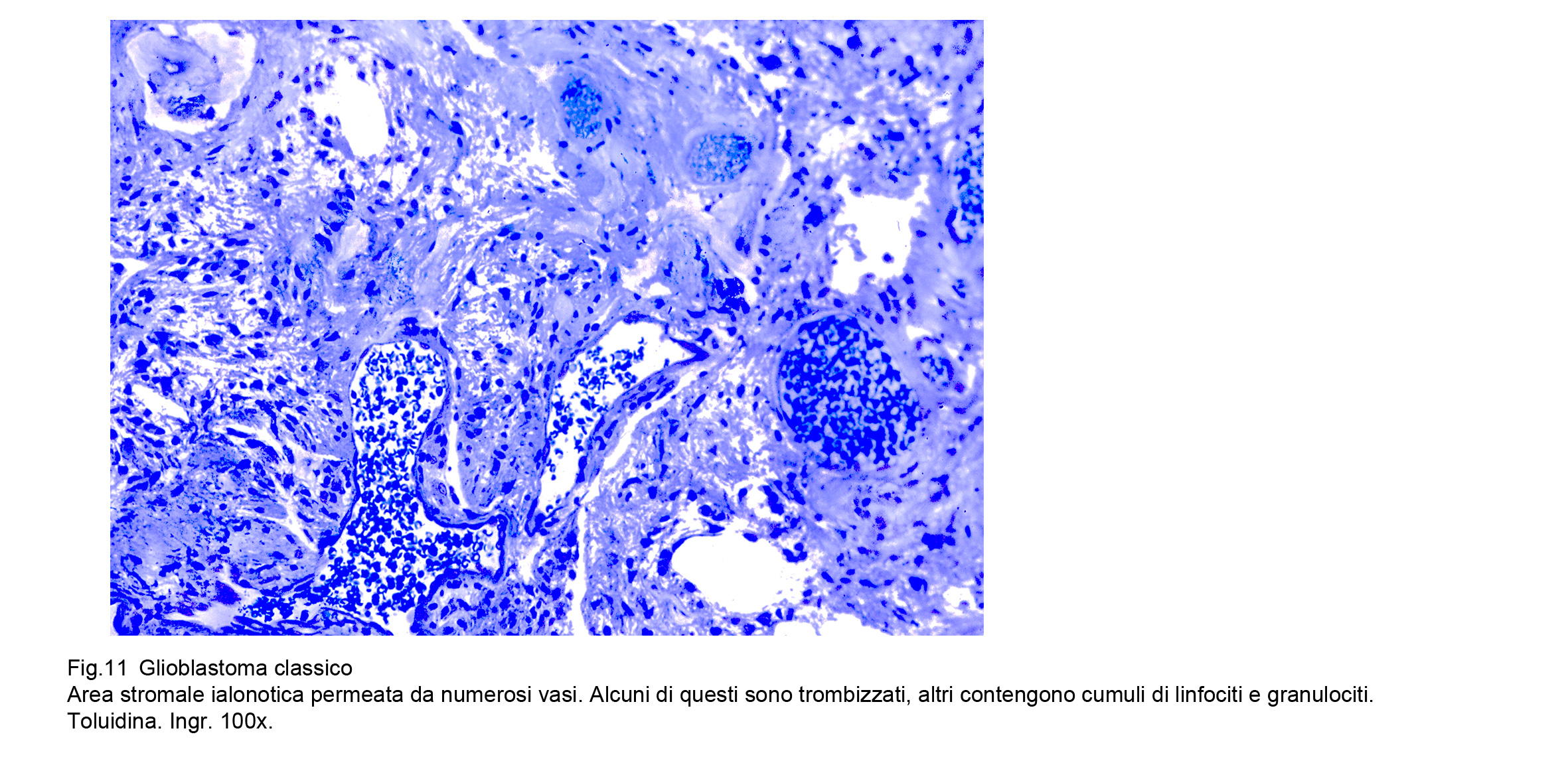
Fig.12 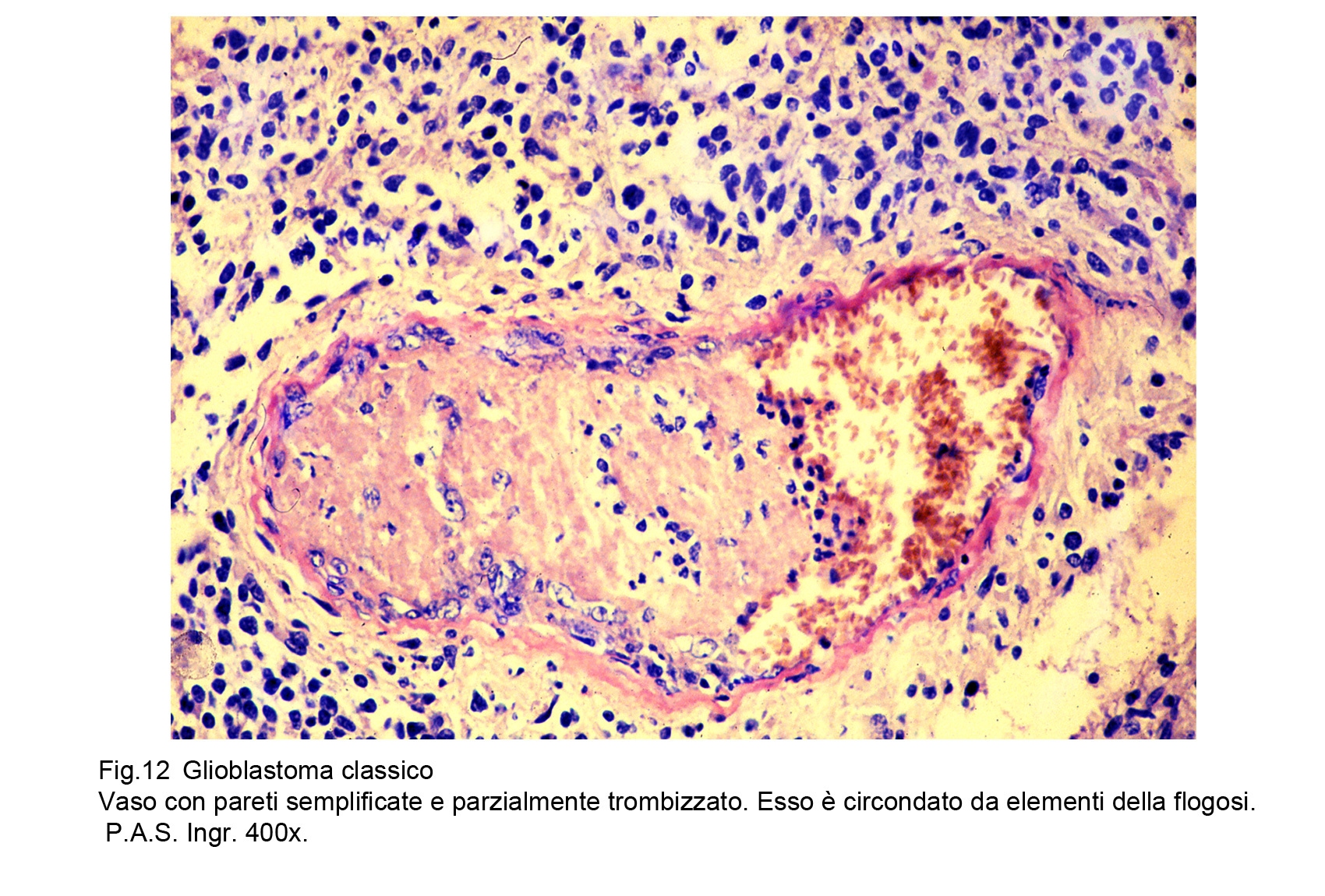 Fig.13
Fig.13 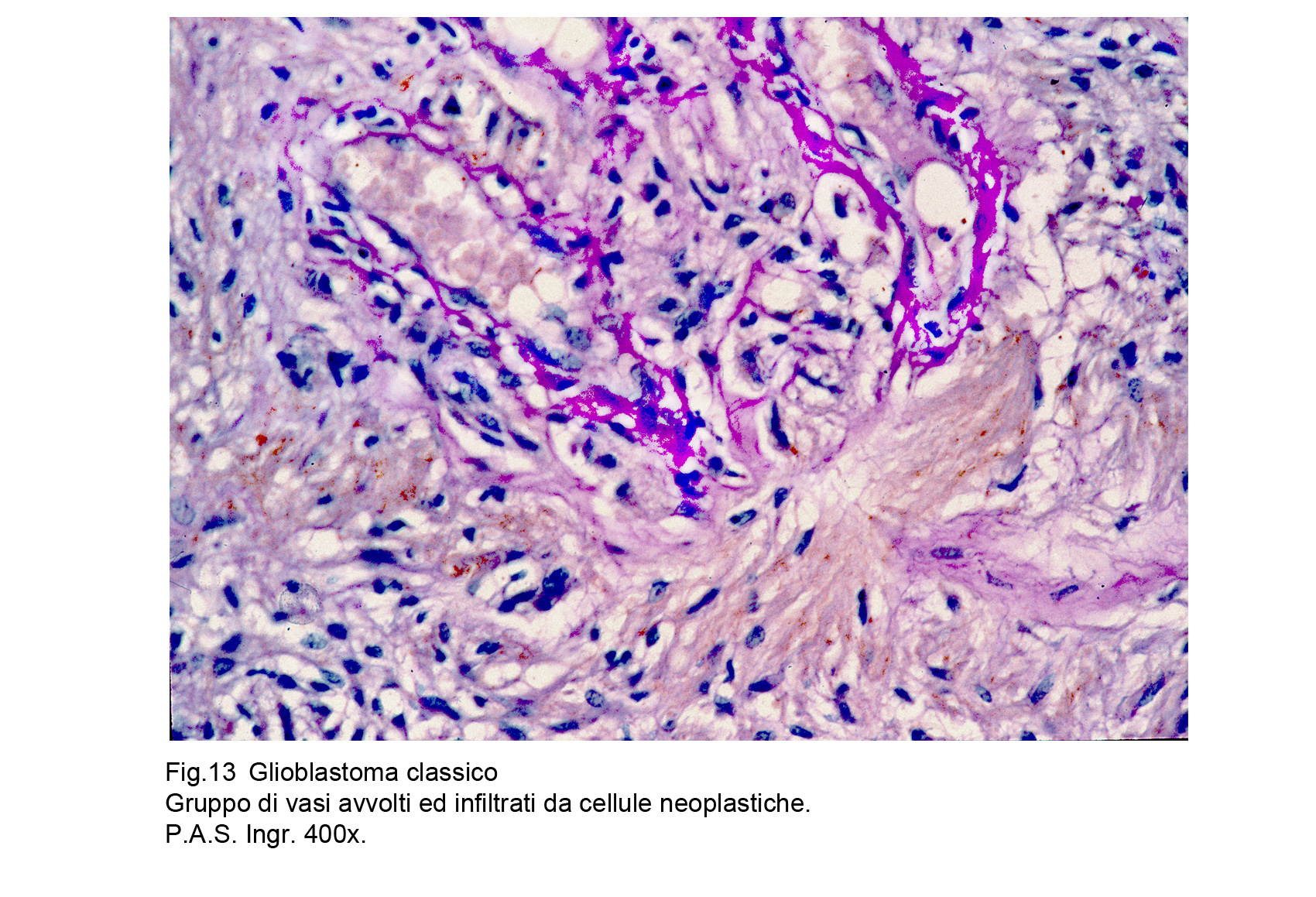
Questo processo di vasculogenesi si realizza con la partecipazione di diversi fattori concausali quali l’ipossia, l’acidosi, l’edema interstiziale e un’azione mediata da molecole sintetizzate dalle cellule neoplastiche. Ricerche di immunoistochimica hanno documentato l’azione mitogena neoangiogenetica mediante i fattori VEGF-2, PDGF, FGF, ANG/TIE, VEGFR-2KDR, l’attività proliferativa degli endoteli mediante le molecole VEF, il grado di permeabilità vascolare medianteil MMP2 e la determinazione dell’ipossia mediante i fattori MMP2 e IHF1.
I GBM sono sede di infiltrati costituiti da elementi immunocompetenti, da macrofagi e dalla partecipazione di microglia su base reattiva.I linfociti, i monociti, i macrofagi si dispongono attorno ai vasi, formando cuffie perivascolari , invadono gli spazi intercellulari e si dispongono a corona attorno ai focolai di necrosi.Le cellule linfo-monocitarie e macrofagiche secernono citochine e sostanze paracrine mediante le quali attivano i processi di fagocitosi e tentano di bloccare o almeno di ridurre la migrazione delle cellule neoplastiche. Indagini condotte a livello molecolare hanno dimostrato che la migrazione di monociti e della microglia è attivata mediante la chemochina CCL5, il fattore VEGF e la neurotensina NTS e hanno evidenziato anche una correlazione tra la espressività del gene CD3G e la risposta immunitaria dei linfociti T. L’entità di questa risposta immunitaria è stata ritenuta importante per l’azione frenante la crescita neoplastica; pertanto è stata tracciata una scala graduata siglata T.I.L. (Tumor Infiltrating Lymphocytes) con la seguente scansione:
Assenza di linfociti,
Presenza di linfociti,
Abbondanza di linfociti.
Questa componente cellulare si arricchisce di una forte reazione microgliale che si rinviene in quantità più elevate sia lungo i margini della neoplasia sia a far barriera lungo il perimetro delle aree di necrosi.Questo insieme di cellule e molecole finalizzato a delimitare la crescita neoplastica è stata indicata con la sigla GAMS (Glioma associated microglia and macrophage),
( Clin. Cancer, Pub.Med. 2013) (Clin. Dev. Immunol, 2013, Pub. Med.)
Le indagini di immunoistochimica per la GFAP hanno evidenziato una buona immunoespressività a livello
delle cellule con un minimo grado di differenziazione e con quote di citoplasma apprezzabili,mentre
hanno svelato una netta negatività per gli elementi indifferenziati; mediante la valutazione di qusto dato
è possibile tracciare una lenea graduata di correlazione tra indice di espressività e differenziazione; alla
stessa maniera,all’inverso, è possibile cogliere rapporti quantitativi di intensità tra ridizione dell’espressività
e processi regressivo-necrotici.
Fig.14 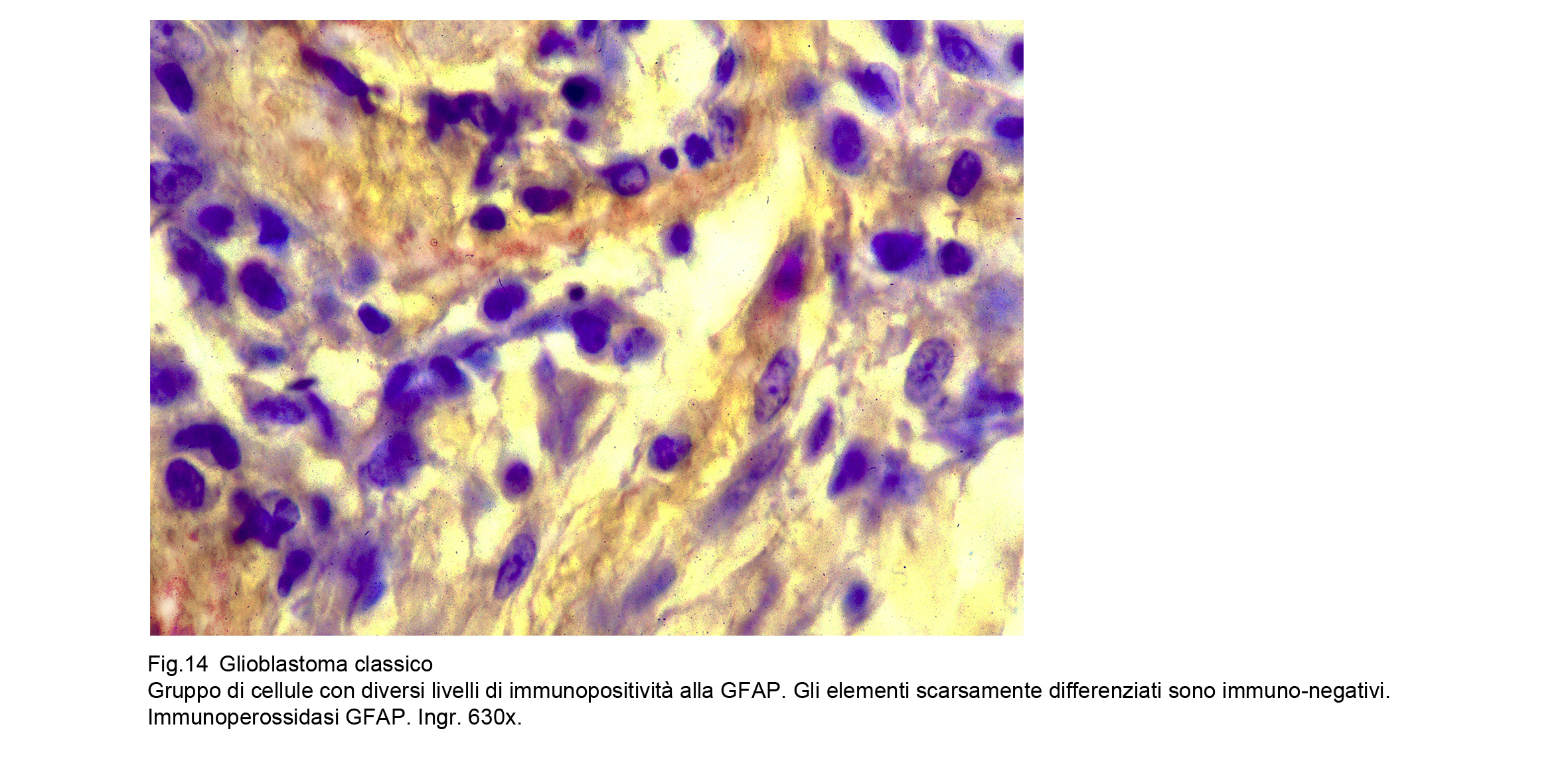 Fig.15
Fig.15 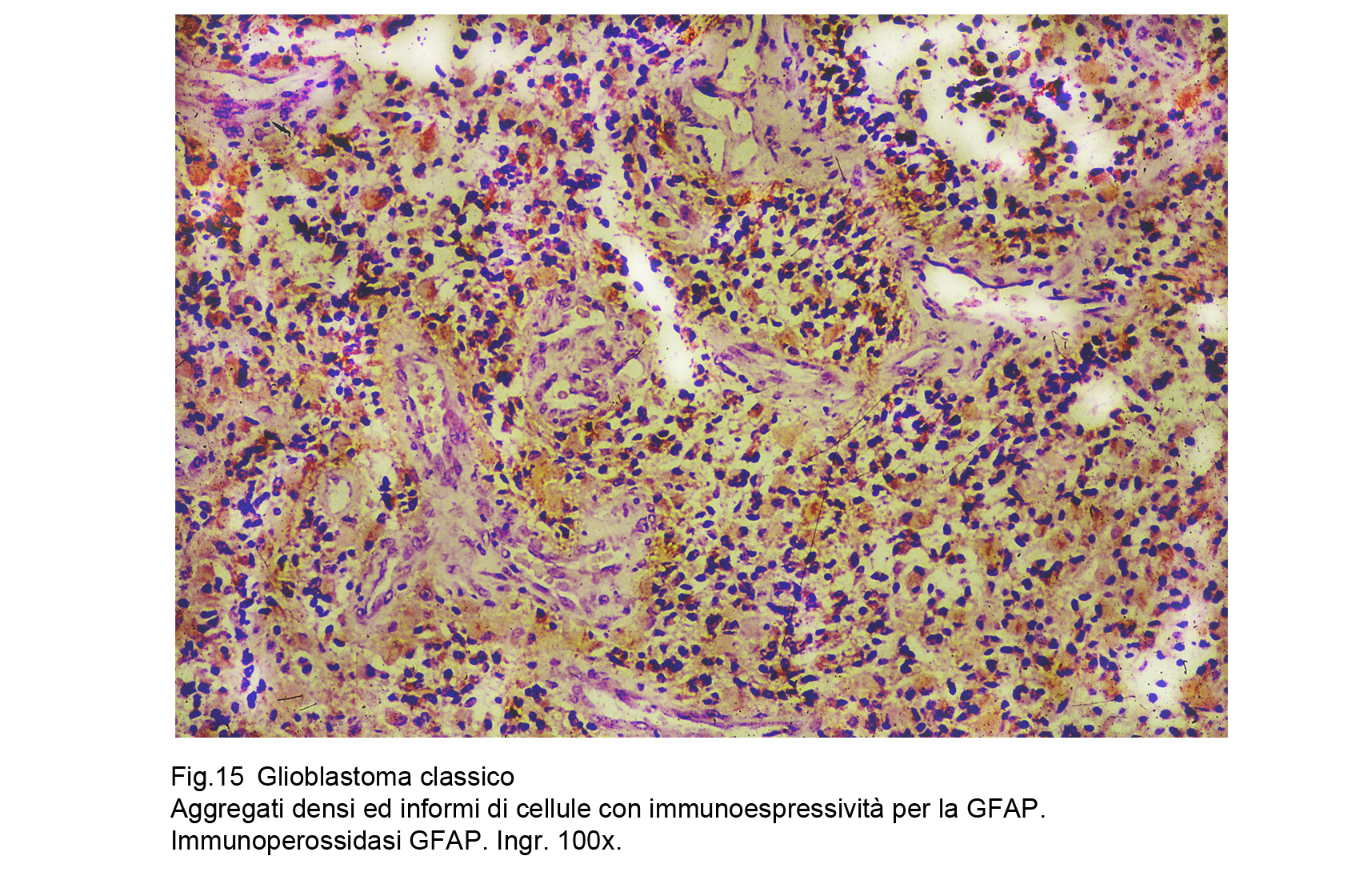 Fig.16
Fig.16 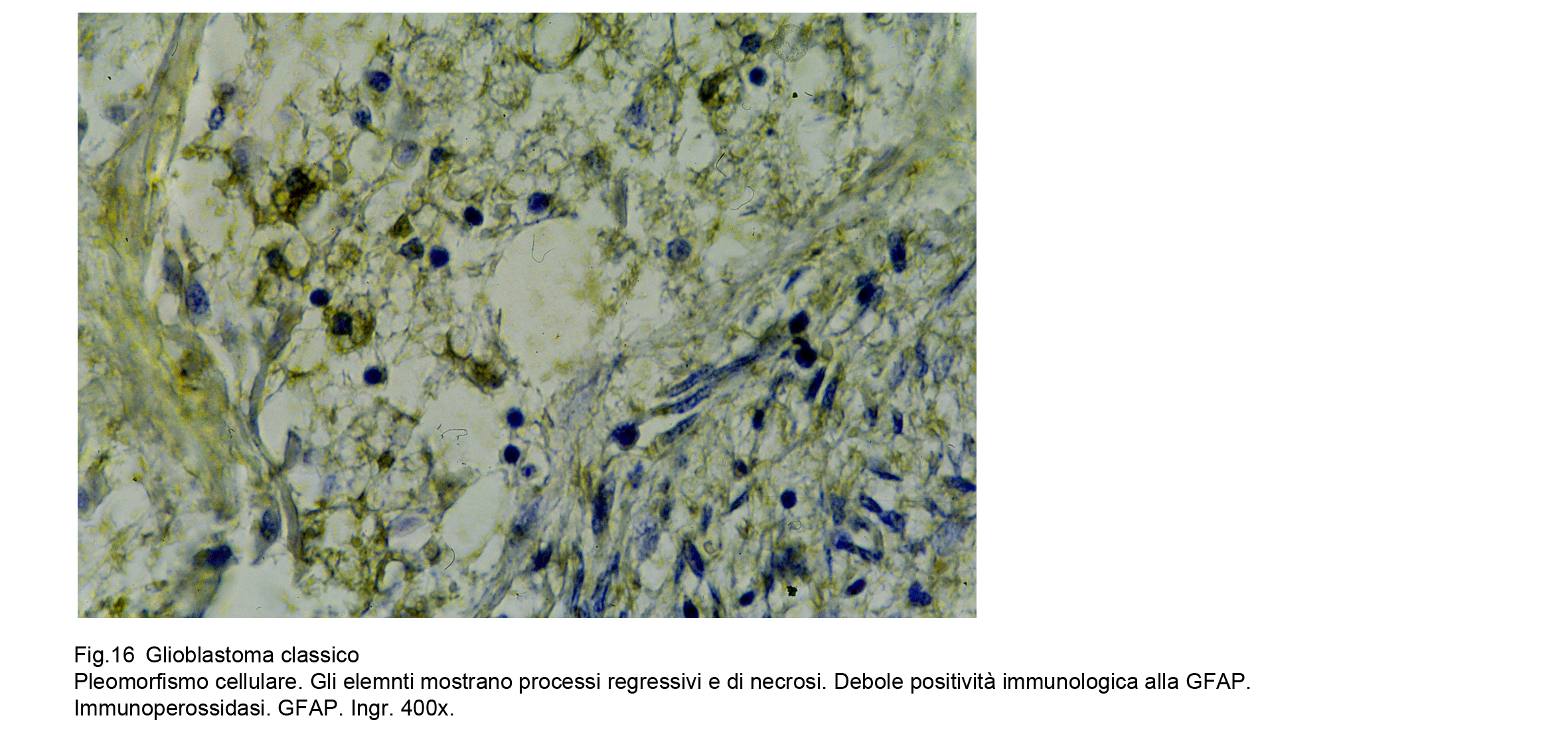
L’esame utrastrutturale al microscopio elettronico a trasmissione ha documentato la esistenza di cellule
coese ma prive di strutture giunzionali , di elementi con scarso citoplasma con organuli in numero ridotto e in preda a rigonfiamento e a vacuolizzazione, e con superfici cellulari sedi di piccoli prolungamenti.
Fig.17 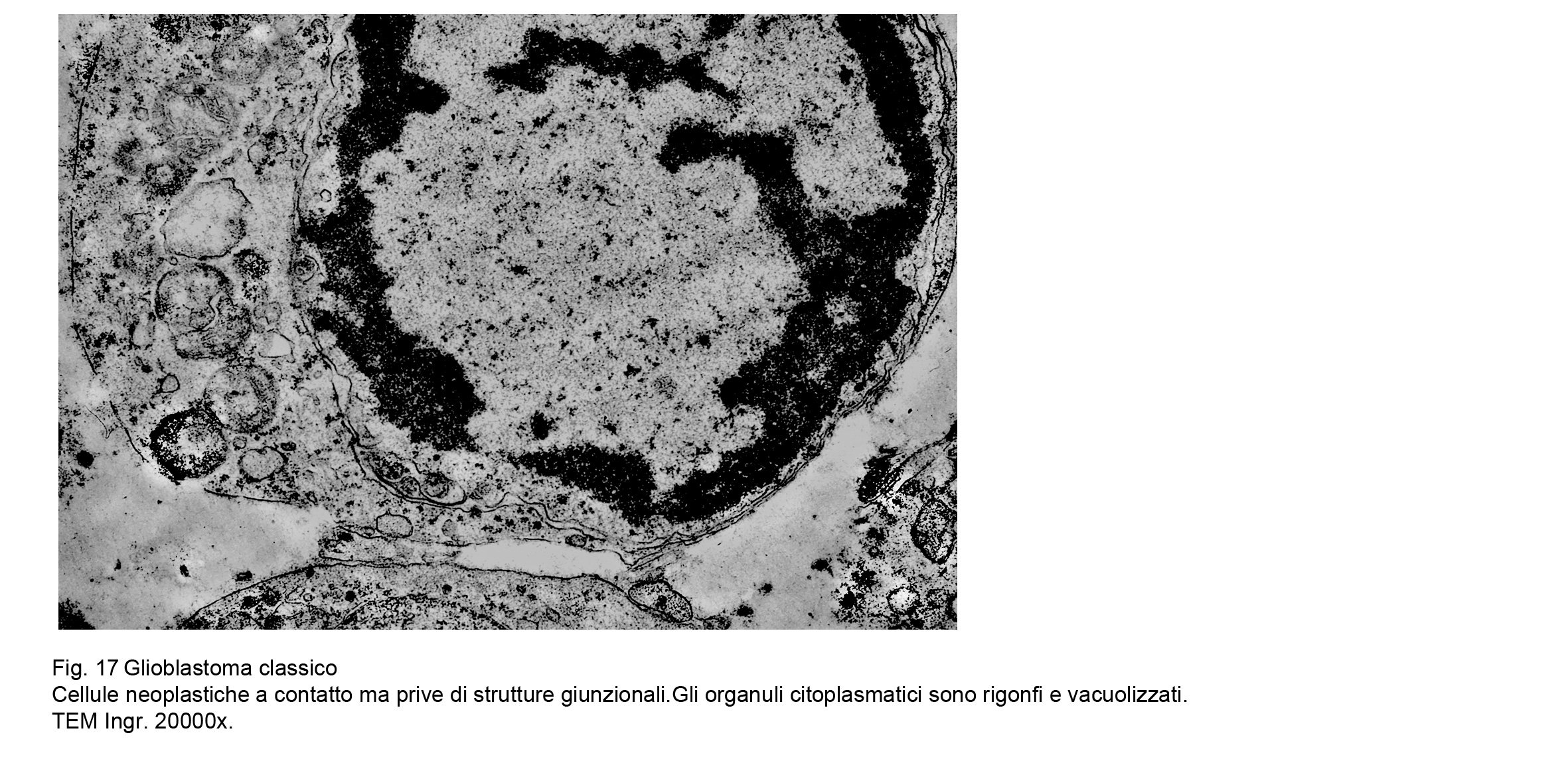 Fig.18
Fig.18 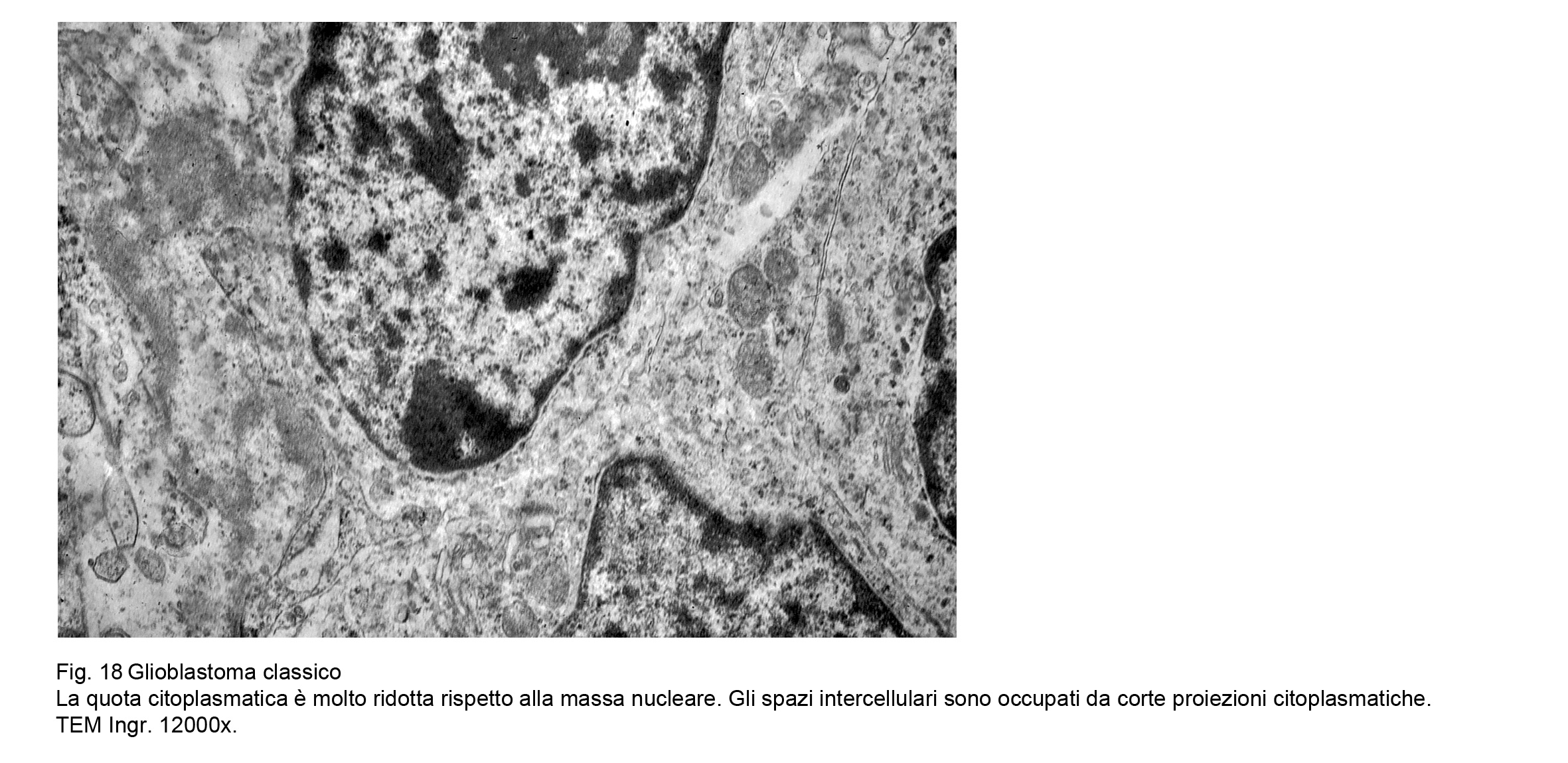
Fig.19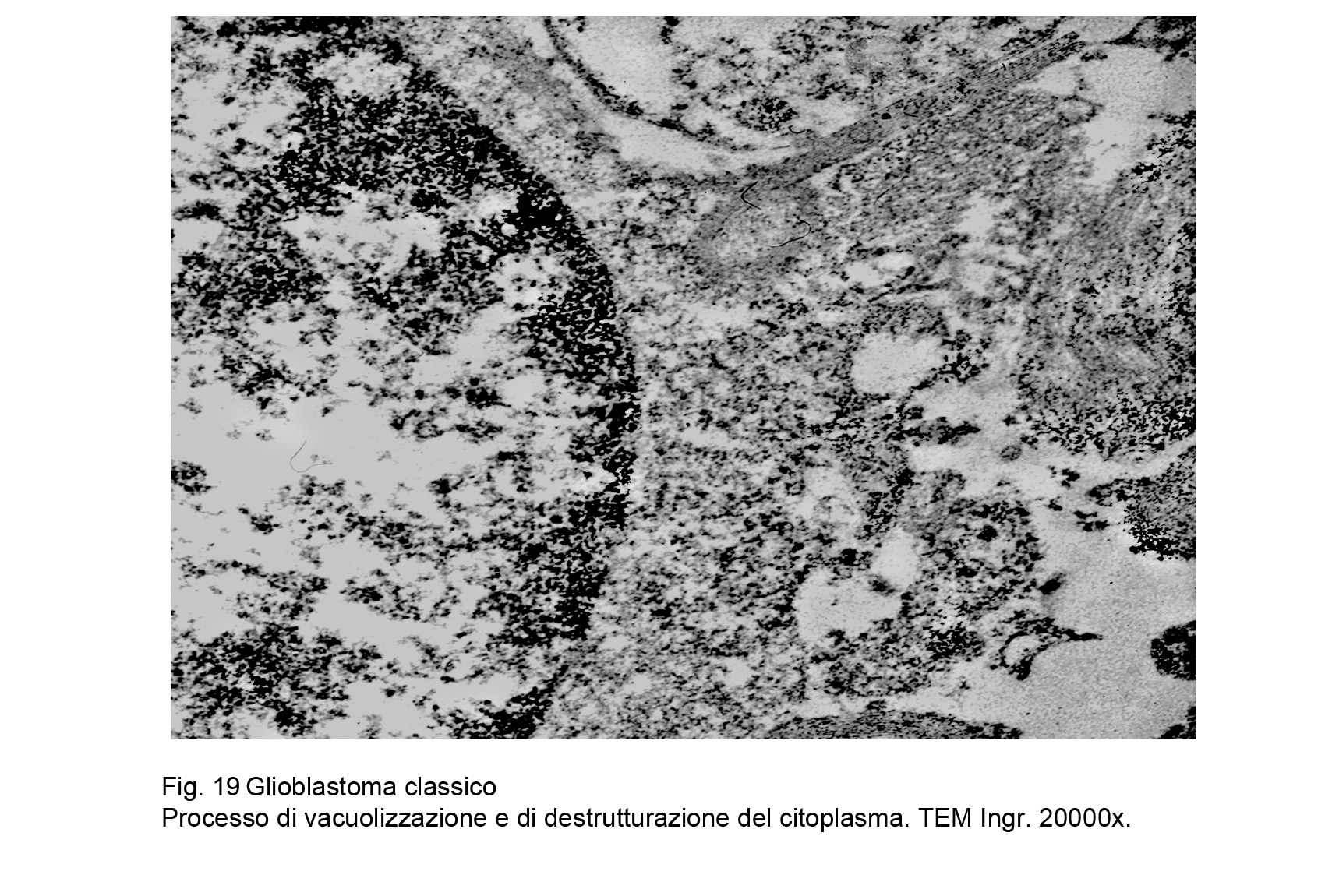 Fig.20
Fig.20 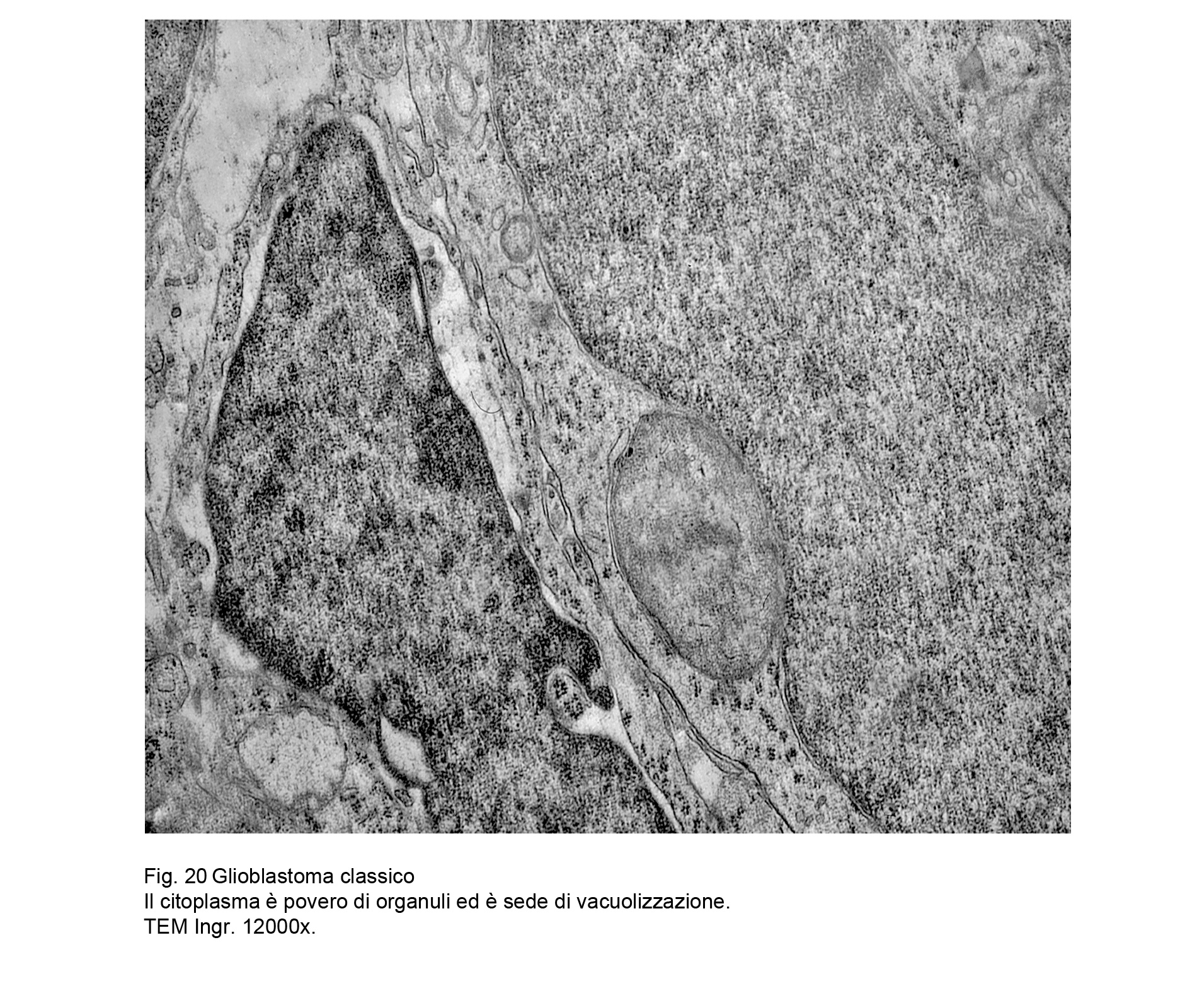
Gliosarcoma
Il gliosarcoma è una variante bifasica del glioblastoma caratterizzata da aree di cellule con differenziazione simil-mesenchimale (WHO).
CARATTERI ISTOPATOLOGICI
Al microscopio luce si evidenzia una componente di natura astrocitaria con caratteri di anaplasia associata alla presenza di aree cellulari aventi i caratteri istopatologici del glioblastoma multiforme. E’ possibile che nell’ambito di tali coacervi cellulari ci siano insule di elementi gliali anaplastici. Accanto a queste si repertano aree occupate da strutture simil-mesenchimali disegnando aspetti di fibrosarcoma,di istiocitoma maligno o aspetti metaplasici di tipo condroide,osteoide o di tessuto mixoide.Queste due componenti (glioblastomatosa e simil-mesenchimale) possono essere distinte e separate oppure ritrovarsi strettamente
commiste e distinguibili solo a livello citomorfologico.
La componente astrocitaria risulta essere immunoespressa per la proteina GFAP, mentre la componente simil-mesenchimale evidenzia una netta immuno-negatività per tale anticorpo.Le indagini genetico-molecolari hanno chiarito la istogenesi della componente simil-mesenchimale cancellando ogni ipotesi del gliosarcoma come tumore da collisione.
Queste indagini sono molteplici e sono riportate qui di seguito:
Mutazione di PTEN, p53, Rb
Delezione p16
Amplificazione EGFR
Mutazione MGMT, IDH1
Alterazione 7,9q, 20q
Rottura dei cromosomi 7p, 9p, 10, 17p e 19p
I risultati delle ricerche su tale assetto genetico hanno accertato che Il profilo genetico è lo stesso sia nelle aree con glioblastomatose sia in quelle aventi i caratteri simil –mesenchimali;questo dato ribadisce la natura monoclonale del gliosarcoma.
GLIOBLASTOMA A CELLULE GIGANTI
il glioblastoma (cgGBM) a cellule giganti appare se è di piccole dimensioni si presenta come una massa circoscritta,ben delimitata, di aspetto compatto, di consistenza sostenuta, di colore grigio-biancastro a sede frequentemente sotto-corticale; viceversa,se questa neoplasia è di grosse dimensioni si rileva che i confini sono poco apprezzabili, il colore è variegato, la consistenza varia da zona a zona e l’aspetto non è uniforme per la presenza di escavazioni di varia grandezza contenenti materiale poltaceo giallastro commisto a sangue.
Al microscopio luce la neoplasia si rivela ipercellulata, pleomorfa per la co-esistenza di cellule giganti bizzarre uni-bi-multinucleate con ampio citoplasma a profilo molto irregolare assieme a elementi di piccola taglia, di forma irregolare, fusata o bipolare. Le cellule, in prevalenza quelle di piccole dimensioni ,sono in attività mitotica tipica ed atipica.
Fig.21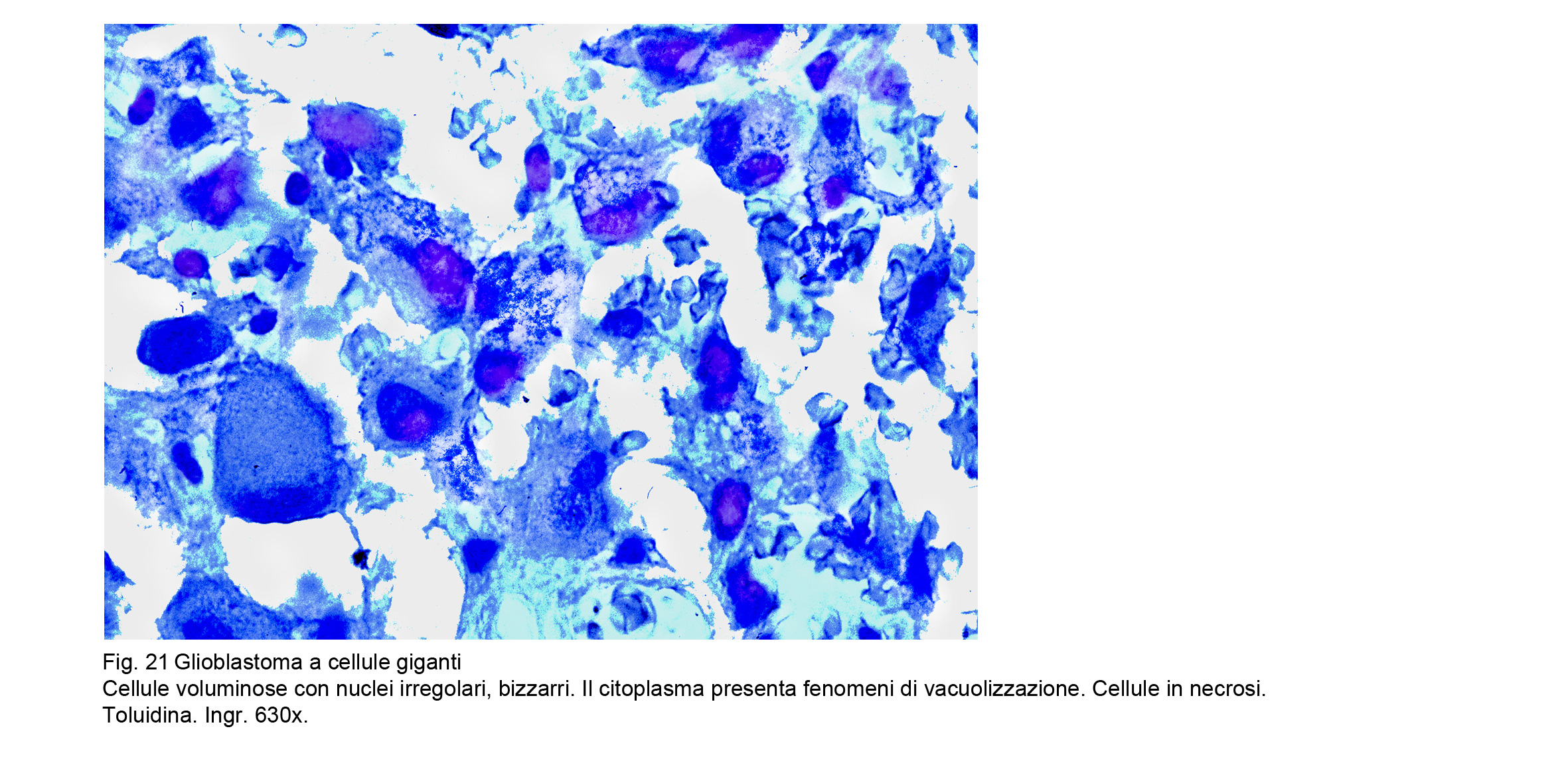
I nuclei sono voluminosi, vescicolosi e contengono un nucleolo preminente. Le cellule sono avvolte e sostenute da uno stroma fibrillare, retiforme, pre-collagene, che si aggancia ai vasi intraparenchimali. Questi sono molto numerosi, sono distribuiti in modo caotico, disordinato, si repertano associati a quote cospicue di capillari. Tali vasi hanno pareti sottili rispetto all’ampiezza del lume e possono essere sedi di ristagno ematico con trombosi oppure lacerazioni delle pareti con spandimento ematico. Molto spesso è dato di riscontrare che i piccoli vasi e i capillari sono circondati da manicotti di linfociti oppure sono compressi ab estrinseco da cellule neoplastiche.
Le indagini di immunoistochimica per la GFAP e per la S-100 hanno evidenziato una marcata immuno-espressività per le cellule sedi di processi di differenziazione,e una immuno-negatività a livello delle cellule indifferenziate
.Il profilo genetico del GBM a cellule giganti è simile a quello del GBM classico. Le cellule giganti mostrano il seguente assetto molecolare.
Poliploidia
Mutazione della p53 e del PTEN
Alterazioni dell’EGFR e p16.
Glioblastoma epiteliale (WHO),Glioblastoma fibrillare- epiteliale (TGCA)
Il glioblastoma fibrillare – epiteliale si distingue per la presenza di una componente simil-epiteliale.Questi elementi si dispongono a formare strutture cribriformi, simil-adenoidee o si organizzano a formare filiere, travate, cordoni cellulari variamente orientati o disposti a rete.
Fig.22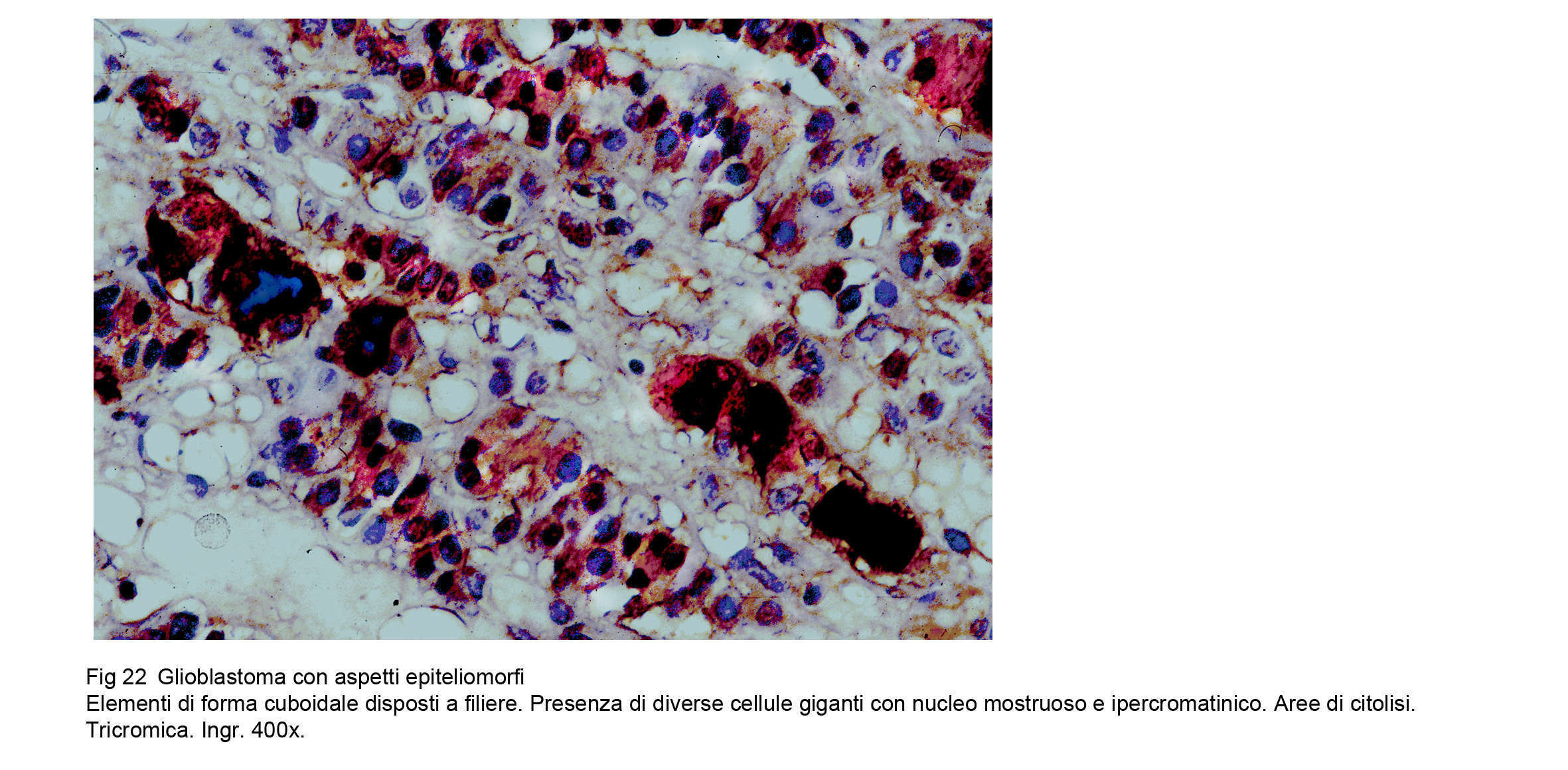 Fig.23
Fig.23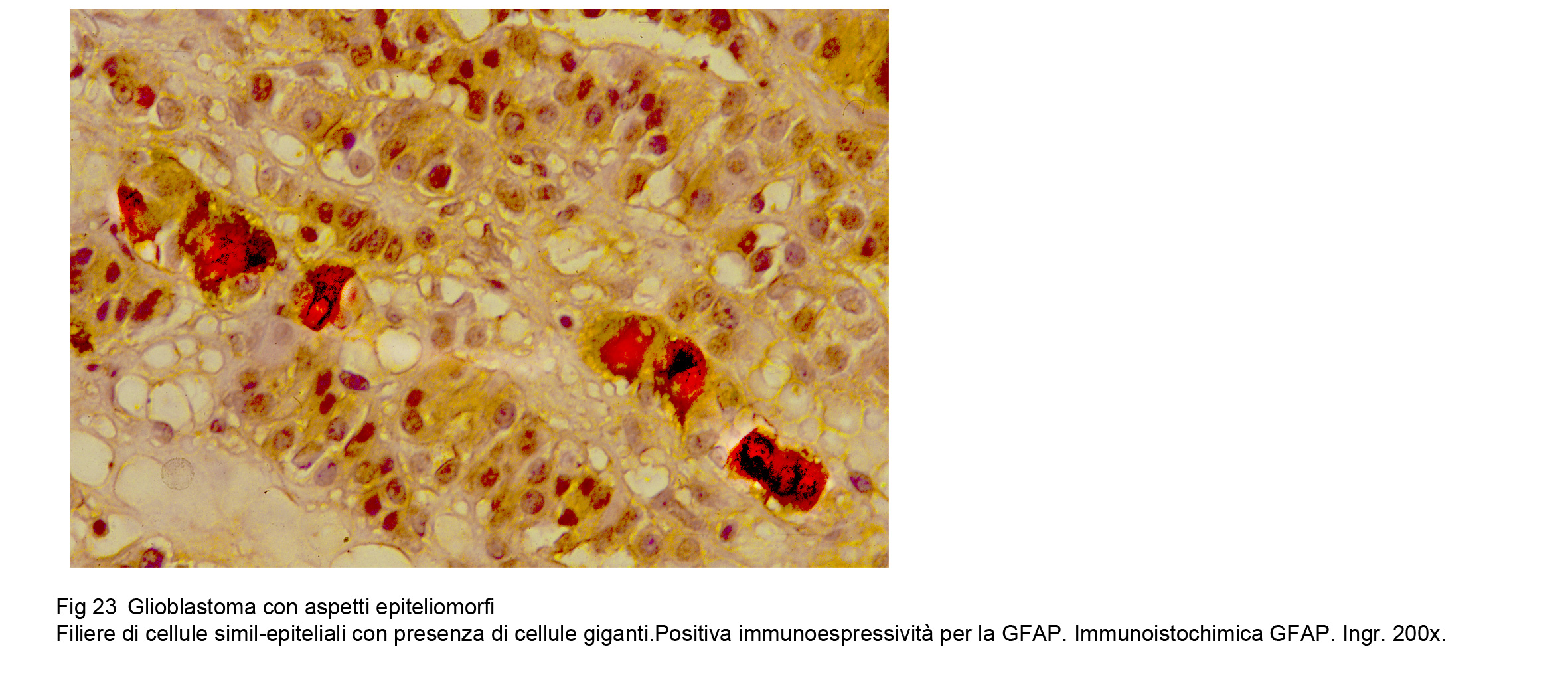
I suddetti reperti istopatologici sono stati integrati dai risultati ottenuti mediante i markers immunoistochimici e da questa operazione sono emerse le seguenti diversità:
- Adenoide-glioblastoma (A-GBM)= Assenza di markers specifici degli epiteli
- Epitelioide – glioblastoma (E-GBM)= Assenza di markers specifici degli epiteli
- Glioblastoma – epiteliale (E-GBM)= Presenza di markers specifici degli epiteli
Le cellule epiteliali si distinguono da quelle epitelioidee perché esprimono una immunopositività per la citocheratina e per l’EMA.
Le ricerche genetico- molecolari hanno documentato le seguenti alterazioni:
Perdita di 1p36, 9p21, 10q23, 17p13, 10q22-26.
Mutazione di EGFR-
(Mol. Pathol, 17, 739-45, 2004),(Clin. Neuropathol, 23, 141-48, 2004).
Glioblastoma a piccole cellue (WHO), Astrocitoma a piccole cellule (TGCA)
La neoplasia è composta da una popolazione cellulare monomorfa, spazialmente disposta in modo diffuso,e pertanto priva di alcun disegno architettonico. Le cellule sono tra loro a mutuo contatto ma prive di coesione : esse sono di forma rotondeggiante, hanno una scarsa quota di citoplasma e sono centrate da un grosso nucleo ipercromatinico, spesso vescicoloso e nucleolato.
Fig.24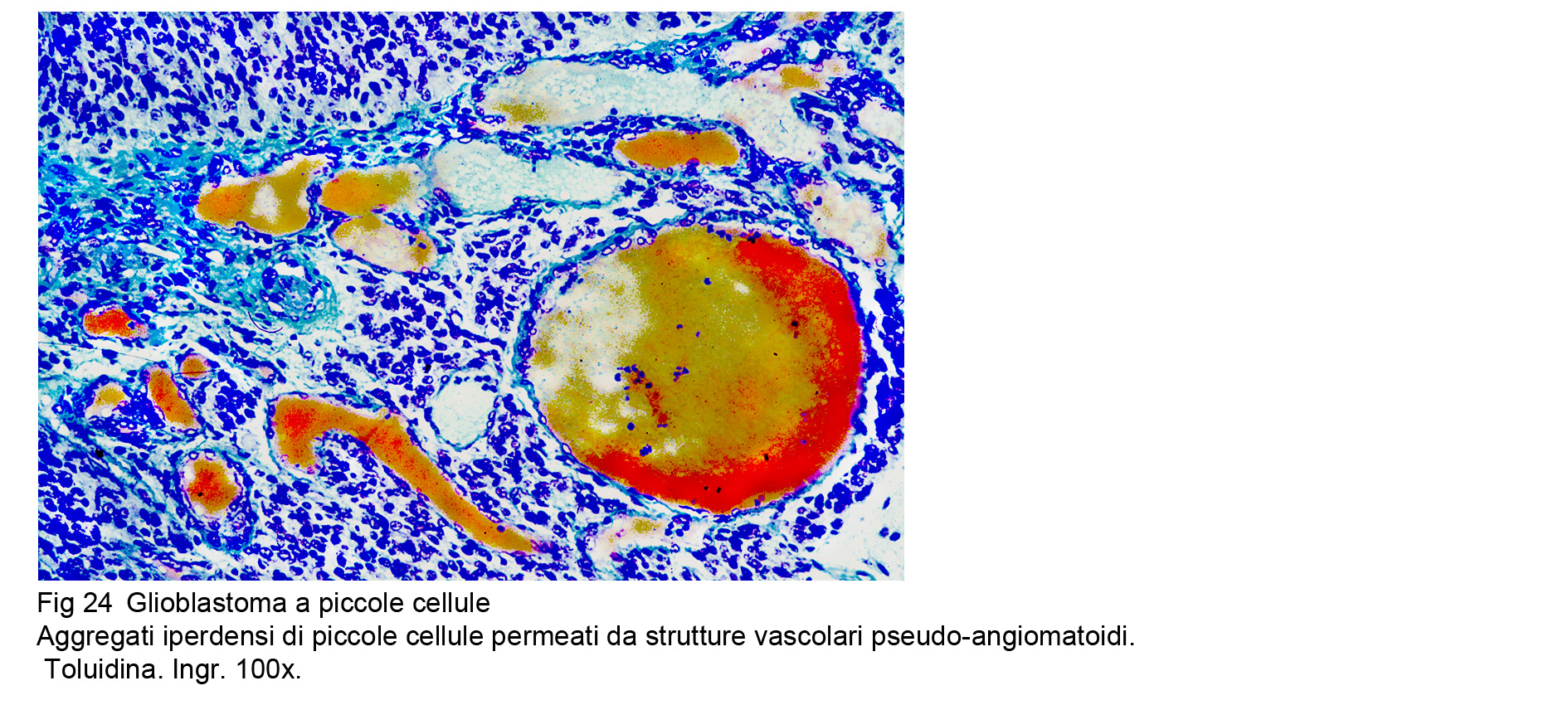 Fig.25
Fig.25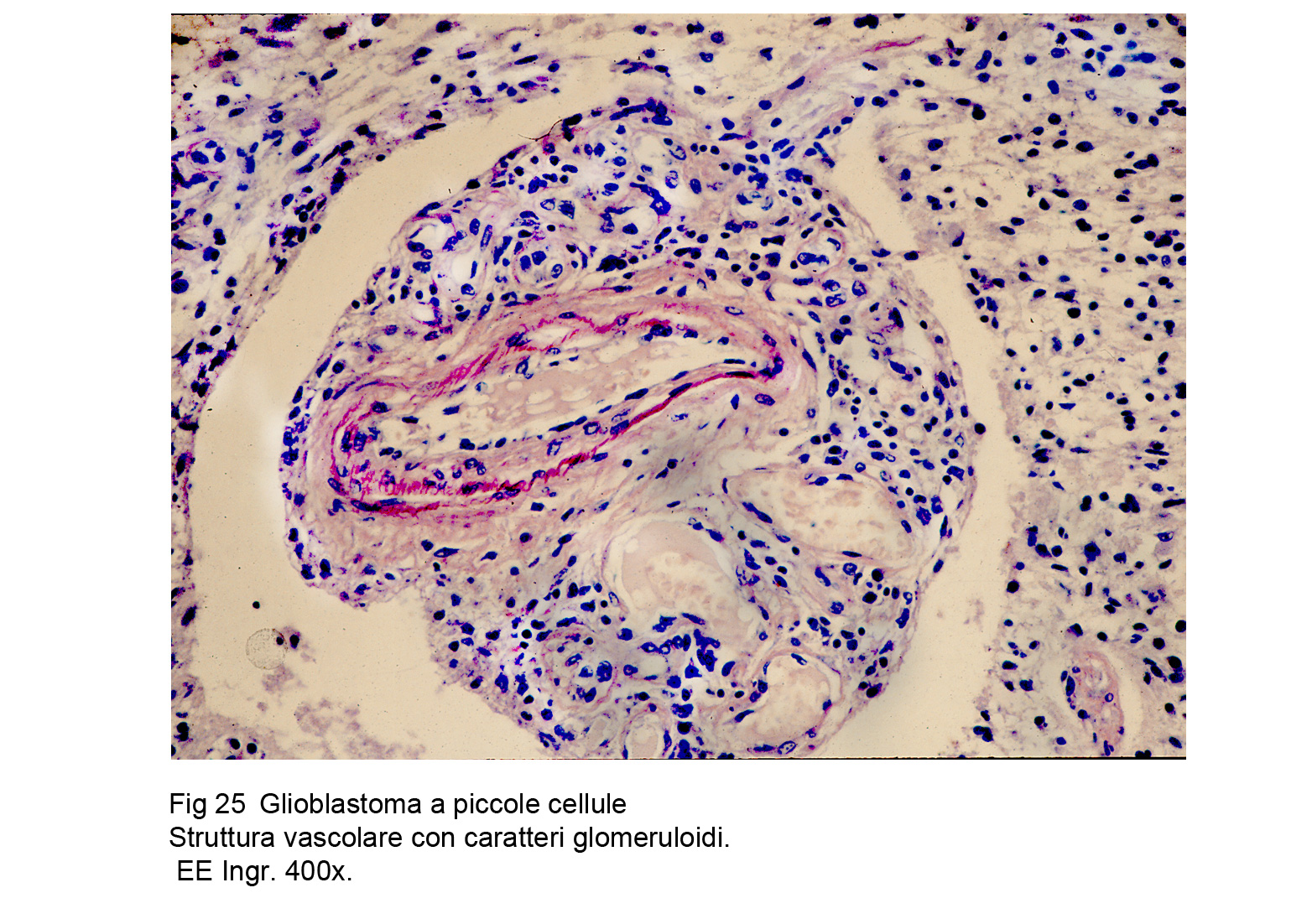 Fig.26
Fig.26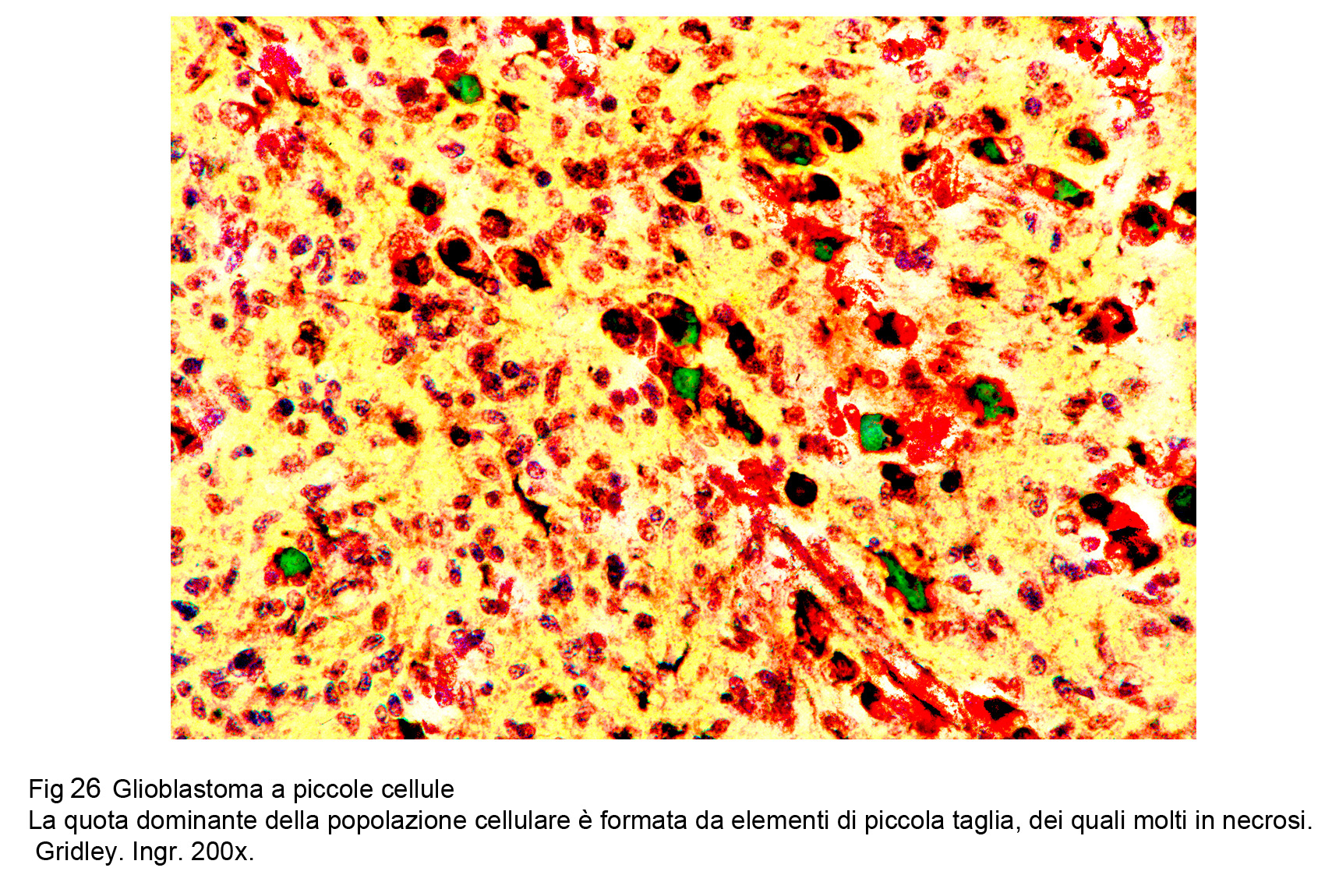
Fig.27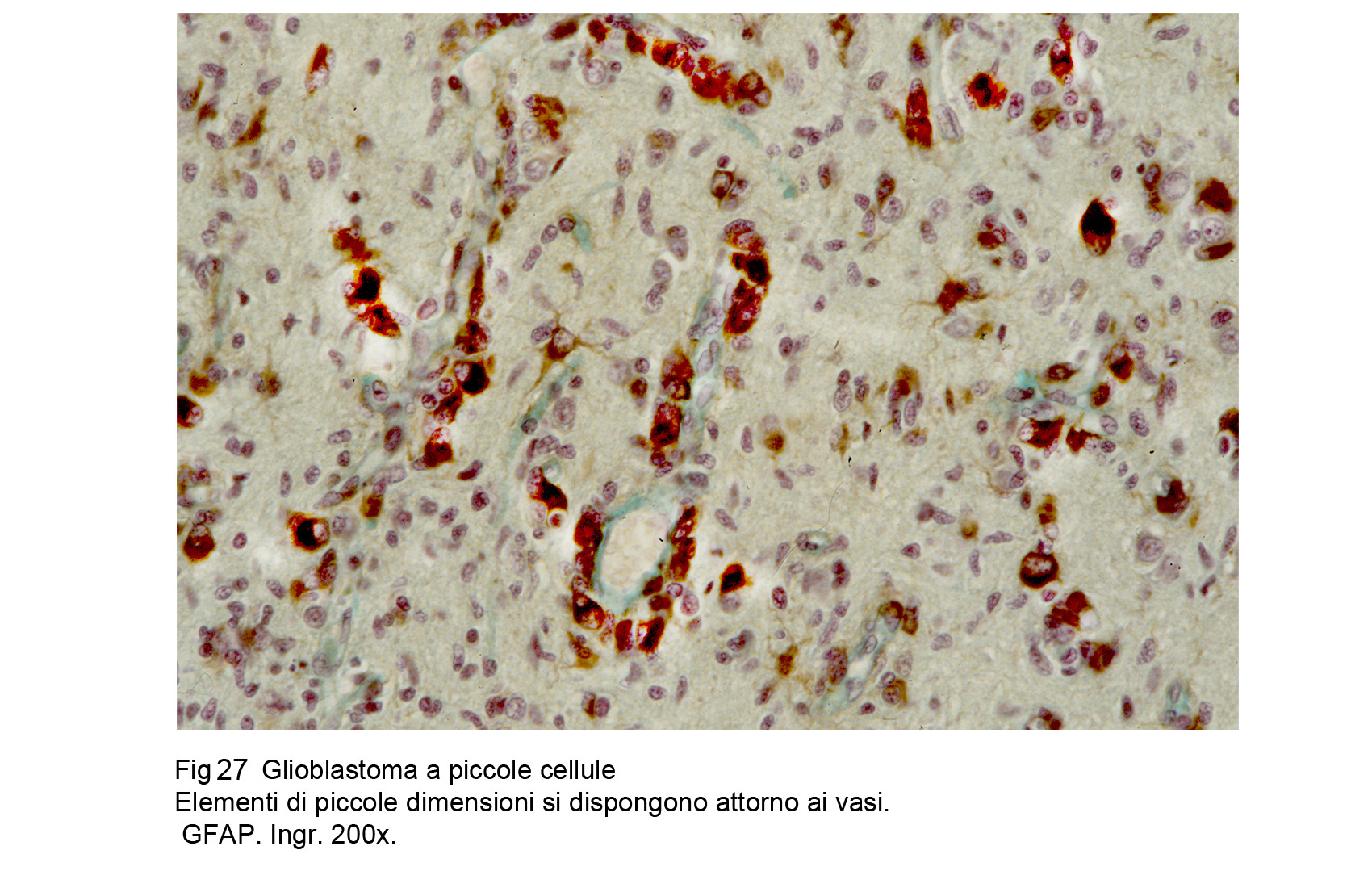 Fig.28
Fig.28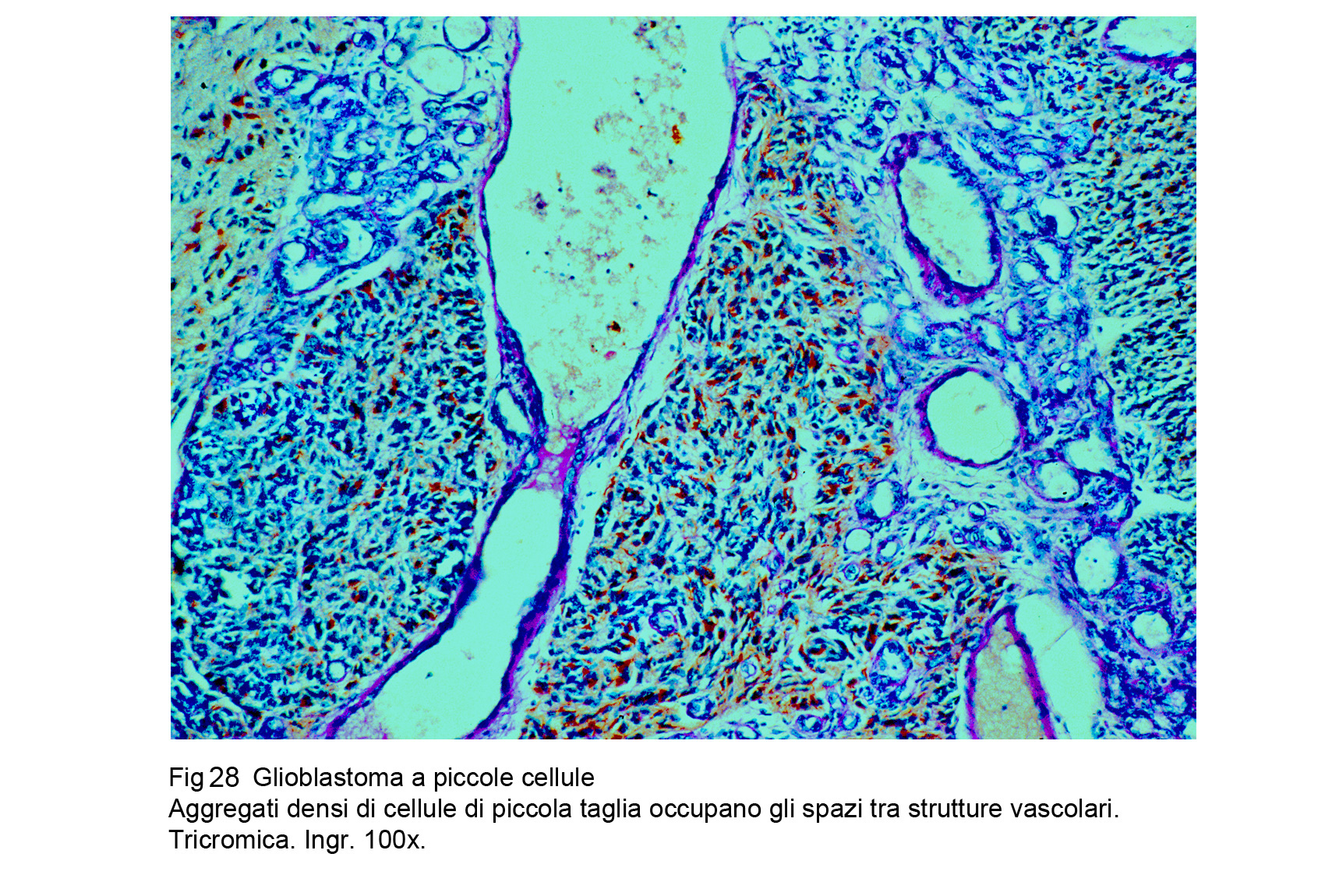 Fig.29
Fig.29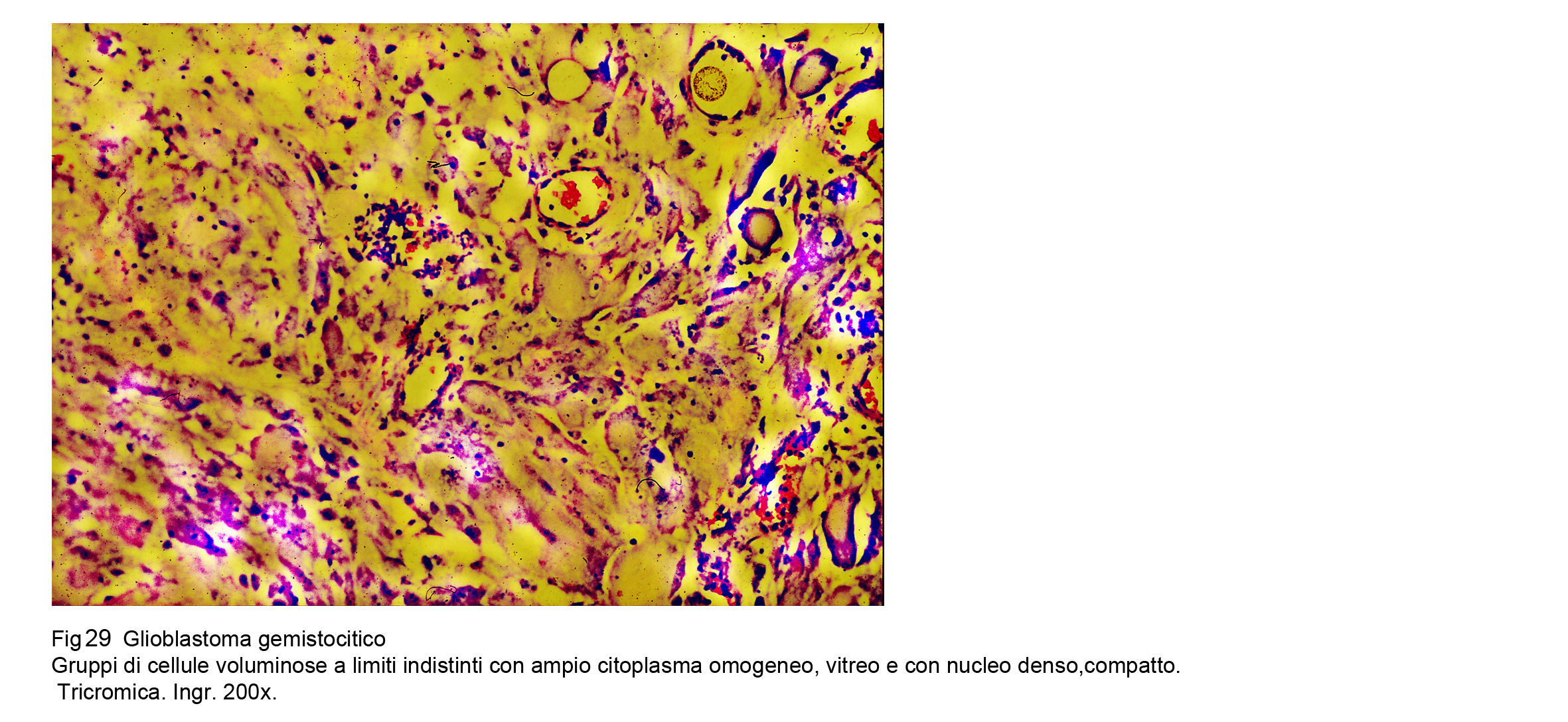
La immunoespressività della GFAP può essere molto labile,tenue e distribuita solo a quote esigue dell’’intera popolazione cellulare.L’indice di proliferazione determinato mediante il MIB-1 è molto alto e si associa alla presenza di molti elementi in mitosi tipica ed atipica.
Il profilo genetico- molecolare risulta essere così disegnato:
Amplificazione EGFR e EGFR VIII
Polisomia del cromosoma 7
Delezione dei cromosomi 10g, 7b, 19q
(Cancer, 101, 2318-2326, 2004) (J. Neuropathol. Exp. Neurol 60, 1099-1104, 2001),(Clin. Neuropath. 24, 163-169, 2005)
Glioblastoma con componente oligodendrogliocitarie (GBMO)
Alcuni casi di GBM contengono in quote variabili aree neoplastiche occupate da cellule oligodendrogliocitiche frequentemente commiste a elementi astrocitari.Se si escludono queste componenti, questi tumori evidenziano i caratteri morfologici e biologici del glioblastoma classico.
L’assetto genetico- molecolaredi questo istotipo risulta essere così costituito:
Mutazione di EGFR ep53.
Ridotta espressività di MGMT e di IDH1.
Perdita di eterozigosità di 1p/199 e109
Su la base dei dati molecolari sono state distinti tre sottotipi:
- GBMO con presenza di cellule astrocitarie
- GBMO con presenza di sole cellule oligodendrocitarie
- GBMO con componente cellulare mista astrocitaria-oligodendrocitaria
- Altro
(Neuro-Oncology 14, 518-25, 2012).(Acta Neuropathol, 123, 841-52, 2012)
Glioblastoma con primitive strutture neuro-ectodermiche (GBM-PNET)
E’ possibile il riscontro di GBM caratterizzato da una doppia componente cellulare, strettamente commista, identificabile sia a livello morfologico sia mediante indagini immunoistochimiche.
La prima quota è formata da cellule glioblastomatose, la seconda è costituita da elementi neuroectodermici (PNET).Infatti, oltre alla presenza più o meno cospicua di cellule del GBM, nei loro multiformi sottotipi, si repertano quote diverse di cellule piccole, rotondeggianti con scarso citoplasma e nucleo sferoidale ipercromatinico; queste cellule si dispongono quasi sempre in ammassi informi, o cordonali con possibile disegno di rosette vere di Homer-Wright.
Le indagini di immunoistochimica confermano la coesistenza di due citotipi: La componente di cellule glioblastomatose presenta una immuno-positività per la GFAP,e la S-100; la quota di cellule PNET rivela una immunopositività per la sinaptofisina,e per la proteina dei neurofilamenti (NFP).
L’assetto genetico-molecolare di questa variante di glioblastoma evidenzia i seguenti caratteri:
Bassa espressività di p53 e PTEN
Mutazione in EGFR, CDK4, MDM2
(Neuropath. Appl. Neurobiol. 28, 325-333, 2002)
Glioblastoma gemistocitico (WHO),Astrocitoma gemisticitico (TCGA)
Questo tumore è formato da cellule astrocitarie gemistocitiche, riconoscibili per il loro ampio citoplasma di aspetto vitreo, omogeneo e per la presenza di un nucleo marginato, denso, ipercromatinico, compatto Queste neoplasie si presentano a livello cito-morfologico quali astrocitomi di II grado,invece a livello biologico-evolutivo si esprimono in modo aggressivo come un glioblastoma. Secondo il WHO, questi sottotipi sono da considerarsi quali glioblastomi con differenziazione in senso gemistocitico.Secondo il TCGA sono da ritenere quali astrocitomi a cellule gemistocitiche con capacità aggressive uguali al glioblastoma. Fig 30
Il TCGA sostiene questa valutazione su la base dei seguenti caratteri genetico-molecolari:
Mutazione p53
Alterazione PTEN, EGFR
Alterazioni dei cromosomi 7 e 10
Iperespressione Bcl-2 e Ki67
(Acta Neuropathol 95, 559-64, 1998),(J. Neuropathol Exp Neurol 59, 679-86, 2000)
Glioblastoma con componente a cellule granulari (WHO),Astrocitoma granulare (TCGA)
Si possono repertare glioblastomi con gruppi di cellule granulose; la quantità di queste cellule è variabile e può essere tale da interessare l’intera neoplasia. Queste cellule mostrano il citoplasma acidofilo ricolmo di granuli, piccoli, sferoidali, PAS positivi.Queste cellule, se molto voluminose rassomigliano a macrofagi ed evidenziano una positività ai marcatori di tali cellule.Sono descritti casi in cui si repertano elementi con aspetti transizionali tra le cellule granulari e neoplasie astrocitiche (WHO). Queste cellule granulari sono riscontrabili in altri gliomi, in meningiomi, in adenomi ipofisari, in diversi tumori di altre sedi.Fig 31
Il profilo molecolare di queste neoplasie a cellule granulari appare così costituito ed è riportato quì di seguito:
Rottura dei cromosomi 1p, 9p, 10q, 17p, 19q
Mutazione di p53, p16, p14 e EGFR
(Brain Pathol 13, 185-94, 2003),(Arch. Pathol Lab Med 132, 1946-50, 2008).
Glioblastoma pediatrico
L’assetto genetico-molecolare dei GBM di pazienti di età infantile si è dimostrato diverso da quello riscontrato in soggetti adulti. Emerge una netta divaricazione tra i caratteri morfopatologici e la identità del patromonio genetico-molecolare.I GBM dell’età pediatrica sono forme primitive,presentano una istomorfologia riferibile in linea di massima al GBM classico ma sono vcaratterizzati da una maggiore instabilità genica con molteplici alterazioni cromosomiche.
I dati genetico-molecolari riportati in letteratura sottolineano i seguenti punti salienti:
Disregolazione meno frequente di MGMJ, IDH1/IDH2, PTEN
Una attivazione più frequente di Ras e AKT
Mutazione di p53
Marcata amplificazione di MDM2 e EGFR
Alterazioni di PDGFRH
Alterazioni nei cromosomi sq, 7, 10q, +39, +16p, 8q, 17p
(J. Clin. Oncol. 25, 1196-208, 2007),(J Clin Oncol. 29, 3999-4006, 2011),(Nature 482, 226-31, 2012)
CLASSIFICAZIONE GENETICA DEI GLIOBLASTOMI
Studi di ingegneria genetica hanno sostenuto esservi la possibilità che i GBM possano derivare da cellule stem di riserva che sono geneticamente mutate.Queste cellule sono caratterizzate da una diversità genomica che non solo è complessa ma anche instabile; in conseguenza di questo stato genetico si manifesta la notevole eterogeneità delle sub-popolazioni cellulari, la varietà del quadro clinico e la difformità delle risposte alle terapie. Ciò è stato documentato mediante metodologie che accertano l’assetto del patrimonio genetico, lo stato dei cromosomi e il grado di regolarità del ciclo cellulare.Le conoscenze acquisite sul campo sono state sistematicamente organizzate e finalizzate a inquadrare la composita realtà nosografica del GBM, a elaborare una classificazione esclusivamente su base genetica e in tale guisa a distinguere nuovi sottotipi, quali entità più aderenti al polimorfo quadro clinico e ai nuovi schemi terapeutici.
La classificazione su base genetica è formata da 4 sottotipi, ciascuno di essi ha un proprio distinto corredo genetico-cromosomiale che si presenta così articolato:
- GBM classico
Amplificazione di EGFR
Alterazione di EGFR III
Mutazione (rara) di p53
Delezione di 9p 21,3
Alterazione del cromosoma 7
Frammentazione del cromosoma 2p
- Proneurale
Mutazione di JDH1 e p53
Amplificazione di 4q12
Alta espressività di PDGFA, NK2, homeobox 2, Oligo2 (geni della crescita oligodendrocitica)
Iperespressività dei geni proneurali SOX, DCX, DLL3, ASCL1
- Mesenchimale
Delezione di 7q11,2 , NF1
Mutazione NF1
Commutazione PTEN, NF1
Espressività di markers mesenchimali CHI3L1, MET) e di markers astrocitari TRADD
Espressività di fattore nucleare (NF-KB) e oncogene virale nomologo B.
- Neurale
Espressività di markers neurali: Neurofilamenti, recettore alfa-1, gamma-amino-butirrico, sinagtogimmina1, polipeptidi light
(Cancer Cell, 17, 98-110, 2010),(Cancer Cell, 24, 331-346, 2013),(Plus One 8, e64169, 2013),
(Frontiers in Biosciences 19, 1065-87, 2014)

