DEFINIZIONE
L’astrocitoma pilocitico è una neoplasia di natura astrogliale relativamente circoscritta, a crescita lenta ma infiltrante; essa è composta da cellule gliali di forma bipolare e contiene fibre di Rosenthal, corpi granulari eosinofili e gocce ialine; può presentare una tessitura compatta, ipercellulata oppure con struttura lassa ipocellulata o ancora una architettura bifasica; essa può essere sede di micro-macrociti (WHO, 2007).
Si localizza a livello infratentoriale, può interessare i nervi ottici, il chiasma, l’ipotalamo, i nuclei della base, il ponte, il cervelletto; meno frequentemente coinvolge gli emisferi cerebrali e il midollo spinale; se si appalesa a livello cerebellare, nell’accrescersi, può riversarsi nella cavità del 4° ventricolo; non è infrequente la possibilità di infiltrare e occupare gli spazi leptomeningei con crescita circoscritta in tale ambito senza diffusione di cellule neoplastiche per via liquorale. Dati statistici mettono in evidenza una maggiore incidenza dell’interessamento leptomeningeo nei casi di astrocitoma pilocitico giovanile.
L’astrocitoma pilocitico, sebbene abbia una propria identità nosografica, mostra nella sua concretezza anatomo-clinica uno spettro di aspetti diversi i quali richiedono approcci terapeutici mirati e valutazioni prognostiche differenti. Una prima diversificazione è quella genetica secondo la quale viene distinto l’astrocitoma pilocitico sporadico da quello associato alla neurofibromatosi tipo 1.
Questo astrocitoma è la neoplasia più frequentemente associata al disordine autosomico dominante causata dalla mutazione del gene NF1 nel cromosoma 17q11.2 (WHO, 2007).
Questa condizione genetica si riflette anche su il discordo clinico per un maggiore rischio di invasione della corteccia cerebellare e degli spazi leptomeningei sia ancora per la dimensione prognostica poiché più facilmente ha la possibilità di deviare in senso maligno o verso il glioblatsoma multiforme. Una seconda diversificazione riguarda l’epoca di vita nella quale si manifesta; questo fattore temporale condiziona la morfologia della neoplasia, il ritorno di crescita e il quadro clinico; pertanto si riconosce l’esistenza di una forma giovanile distinta da quella dell’adulto.
La terza diversificazione si ritrova nella variante indicata con il termine di astrocitoma palomixoide; questa variante si distingue a livello morfologico, a livello genetico e a livello clinico per la sua aggressività biologica. Una quarta diversificazione è di tipo istomorfologico; essa è espressa dalla coesistenza di cellule fusate, bipolari dell’astrocitoma pilocitico con quote diverse di astrociti fibrillari, genistocitici e/o oligodendrociti. Questo bi-morfismo o polimorfismo citologico può essere superato, per una diagnosi di certezza, solo mediante metodiche di genetica molecolare. Questo complesso stato di espressività anatomo-cliniche impone analisi dettagliate e coordinate di morfologia, immunoistochimica, ultrastruttura e genetica molecolare.
CARATTERI MACROSCOPICI
L’astrocitoma pilocitico, all’esame macroscopico, appare come una neoformazione di dimensioni variabili, di colore grigio, circoscritta ma priva di una capsula di contorno, e di consistenza soffice o molle.
Sezionando in modo seriale la neoformazione si evidenzia la presenza di aree compatte commiste ad aree spongiosiche e alla esistenza di micro-macrocisti; da queste defluisce liquido trasparente, opaco, di colore giallo rossastro. Questa descrizione di base, nella concretezza dei singoli casi, subisce variazioni in rapporto ai caratteri istopatologici predominanti (aspetto compatto o aspetto lasso), in rapporto alla aggressività biologica (istotipo di astrocitoma pilocitico o astrocitoma pilomixoide), presenza di processi di anaplasia e ancora fenomeni secondari occasionali e subentranti quali edema, emorragie, necrosi, depositi di emossiderina, focolai calcifici, processi di fibro-ialinosi dei vasi.
CARATTERI ISTOPATOLOGICI
La descrizione al microscopioluce si articolainizialmente secondo una visione strutturale d’insieme e successivamente si sviluppa secondo analisi dettagliate.
Seguendo questo metodo di osservazione si rileva che l’architettura d’insieme dell’ astrocitoma pilocitico varia da caso a caso per la esistenza di una struttura composita quanto variabile di aree compatte, zone spongiosiche coesistenti con escavazioni micro-macrocistiche.
Le aree compatte sono formate da aggregati iperdensi di cellule disposte in modo diffuso o zonalmente cordonale; tale popolazione cellulare è attraversata da numerosi vasi ed è sepimentata da tralci fibrotici.
Fig.1 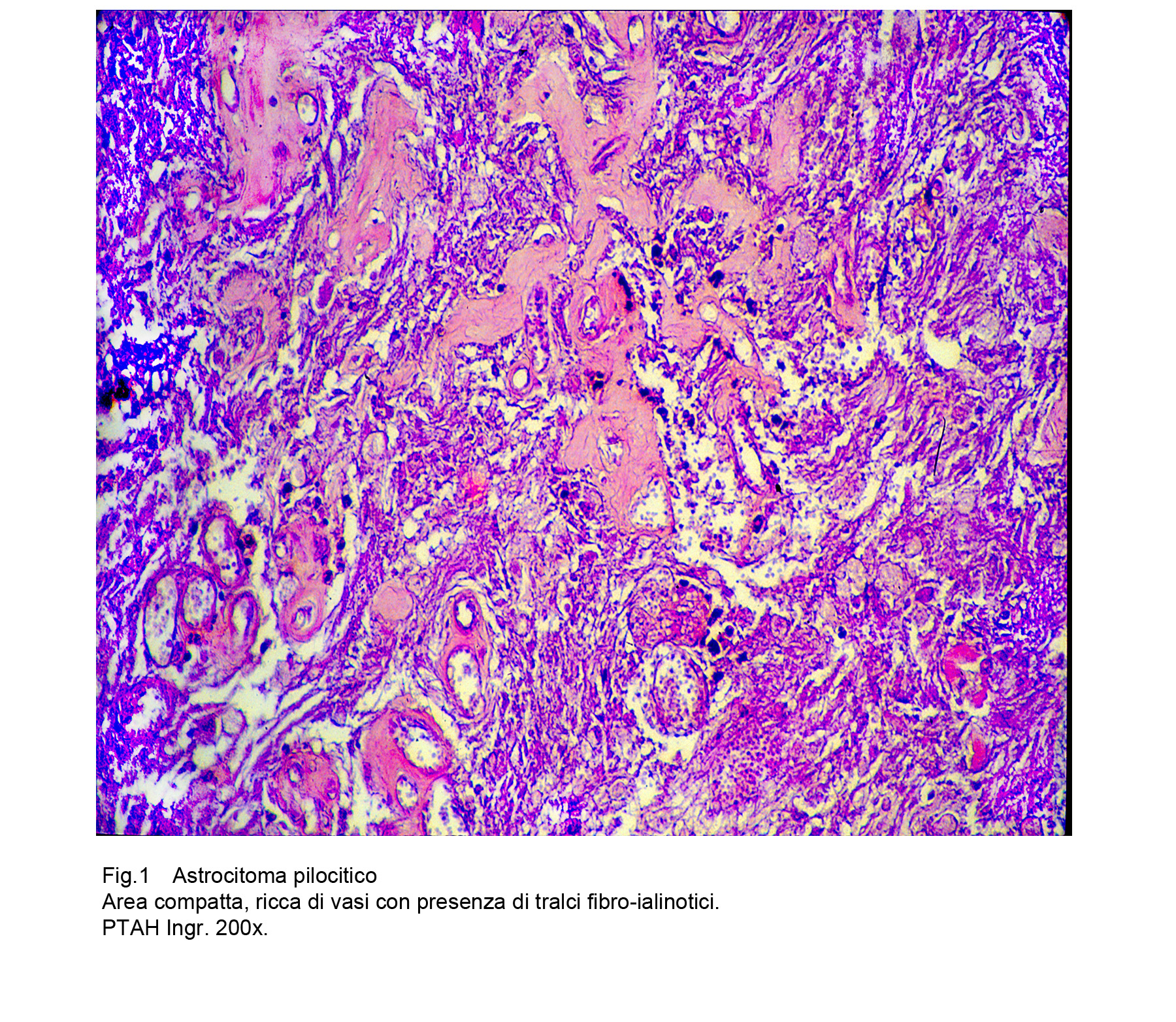
Le zone spongiosiche hanno una struttura lassa per la presenza di ampi spazi intercellulari e mostrano una popolazione cellulare ridotta o addirittura esigua.
Fig.2 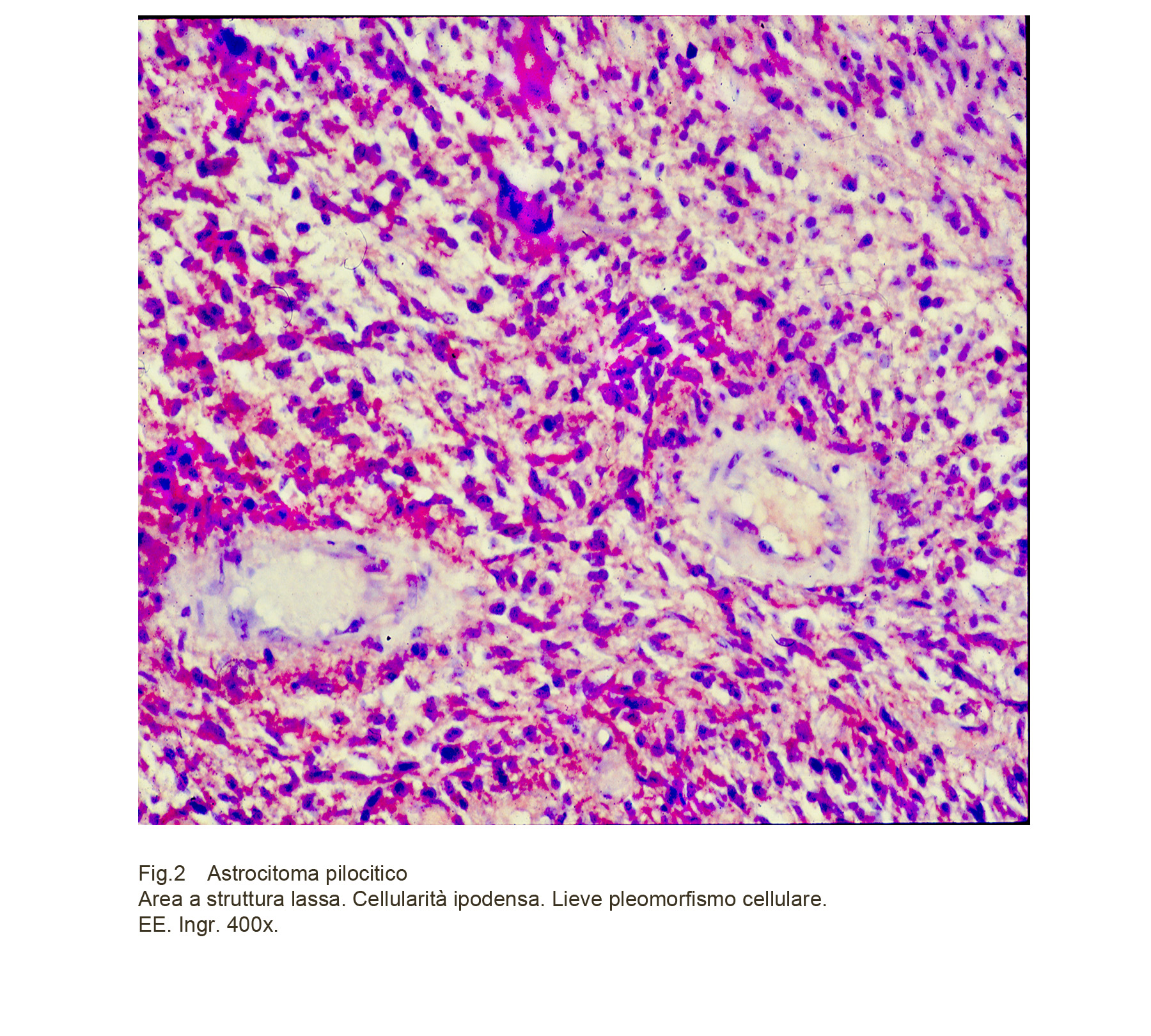
Nella maggior parte dei casi, l’astrocitoma pilocitico è costituito dalla copresenza, disposta in modo casuale e quantità variabile di aree compatte e di aree spongiosiche, acquisendo così l’appellativo di astrocitoma pilocitico bifasico.
Fig.3 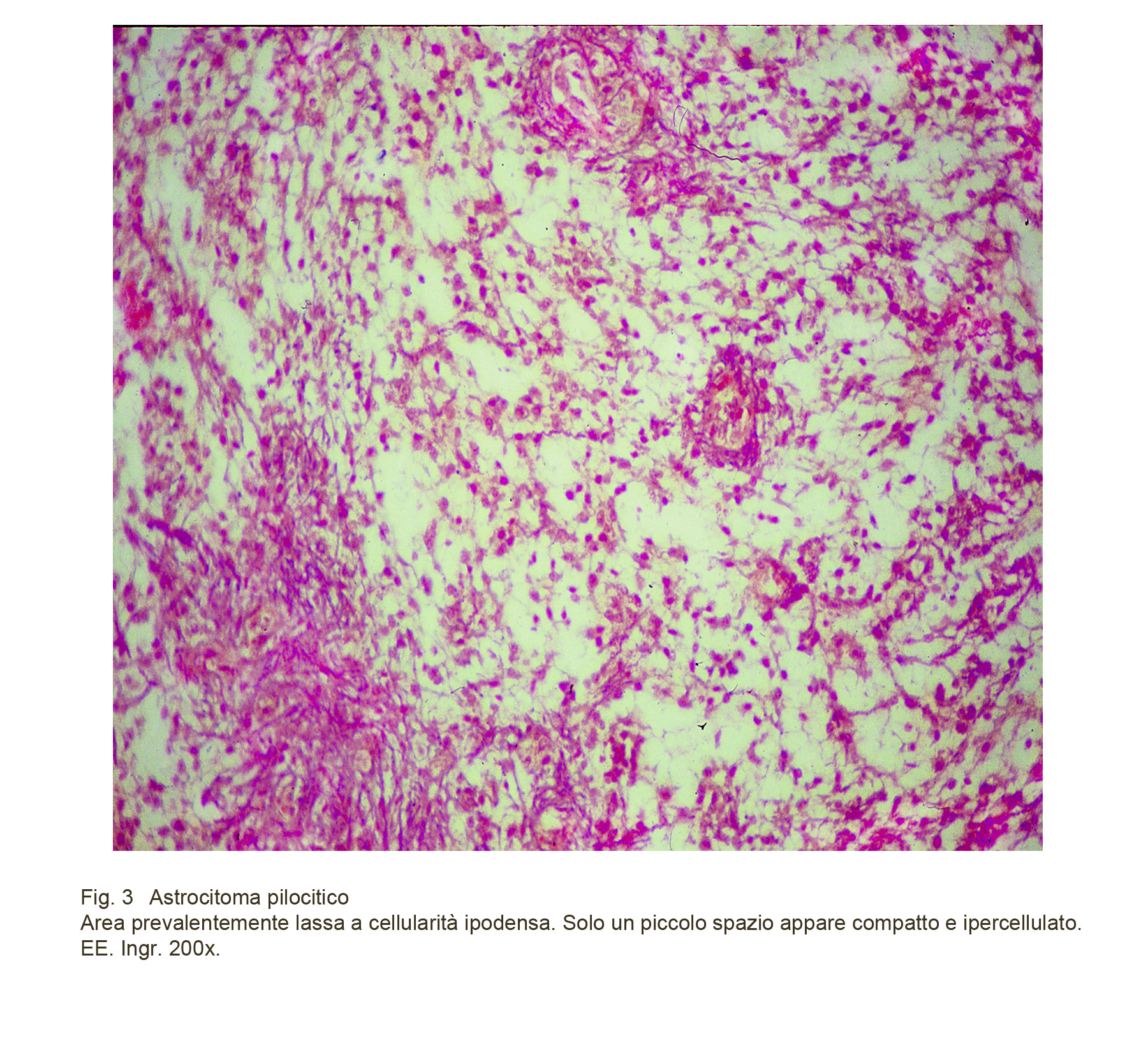
A livello delle aree spongiosiche il processo di rarefazione delle quote cellulari può essere progressivo e plurifocale; di conseguenza si formano, nel tempo, piccole escavazioni prive di pareti prioprie che, coalescendosi, si trasformano in macro-escavazioni, impropriamente denominate cisti.
Fig.4 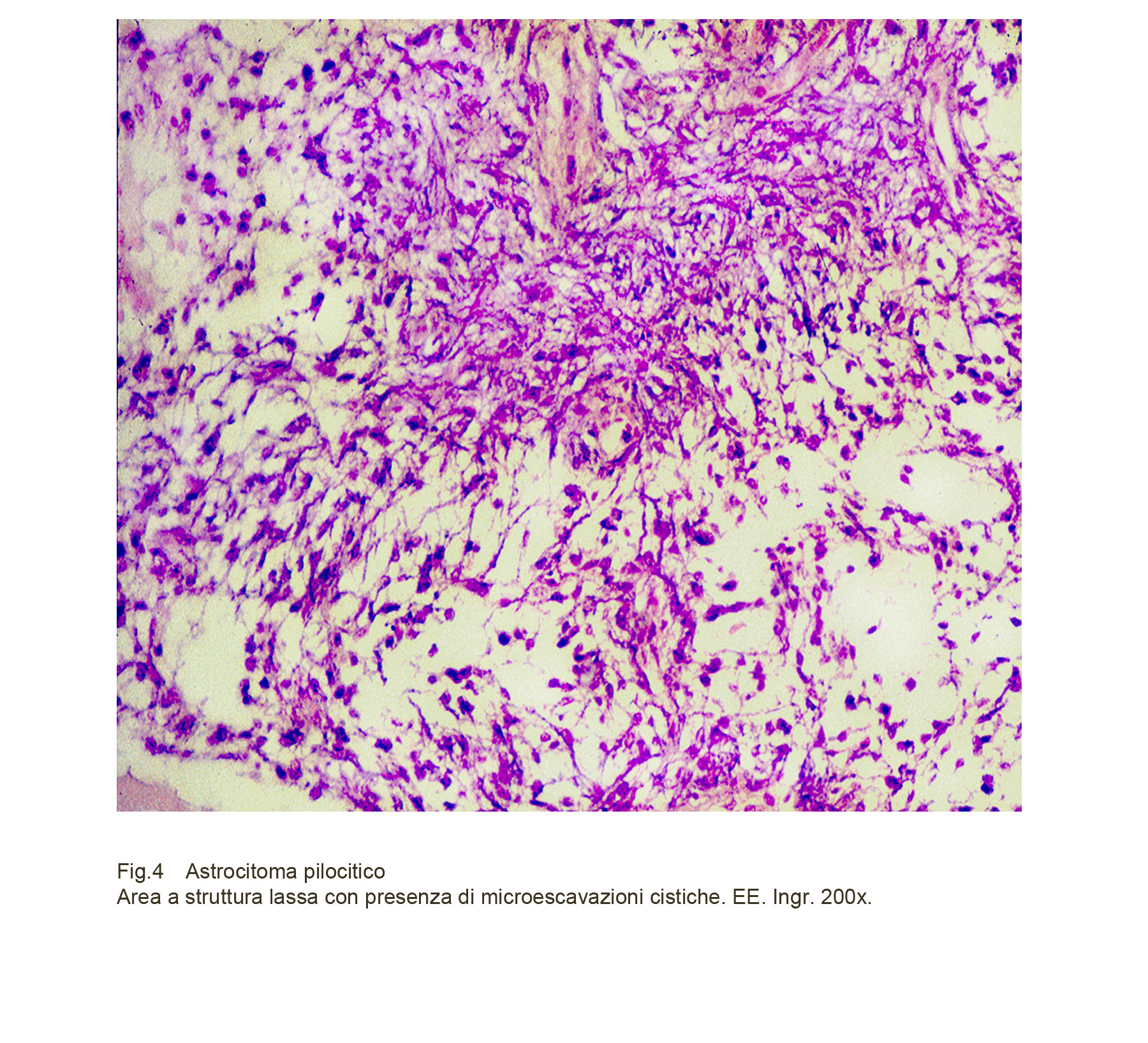
Successivamente a questa breve descrizione d’insieme è opportuno passare a una analisi dettagliata di questa neoplasia. Nelle aree compatte il reperto maggiormente eclatante e più significativo è dato dalla presenza di una popolazione cellulare di media taglia, di forma fusata, di aspetto bipolare. Da queste estremità polari si dipartomo prolungamenti glio-fibrillari sottili, di varia lunghezza; essi si congiungono e si intrecciano con quelli delle cellule vicine tracciando così un disegno a rete di densità variabili. Questi elementi sono tra loro a mutuo contatto ma privi di strutture di coesione intercellulare e sono riuniti in sottili fasci mono-direzionali. Queste cellule sono monomorfe, hanno scarso citoplasma eosinofilo e sono fornite di nucleo ovoidale o allungato, normocromatinico apparentemente privo di nucleolo.
Fig.5 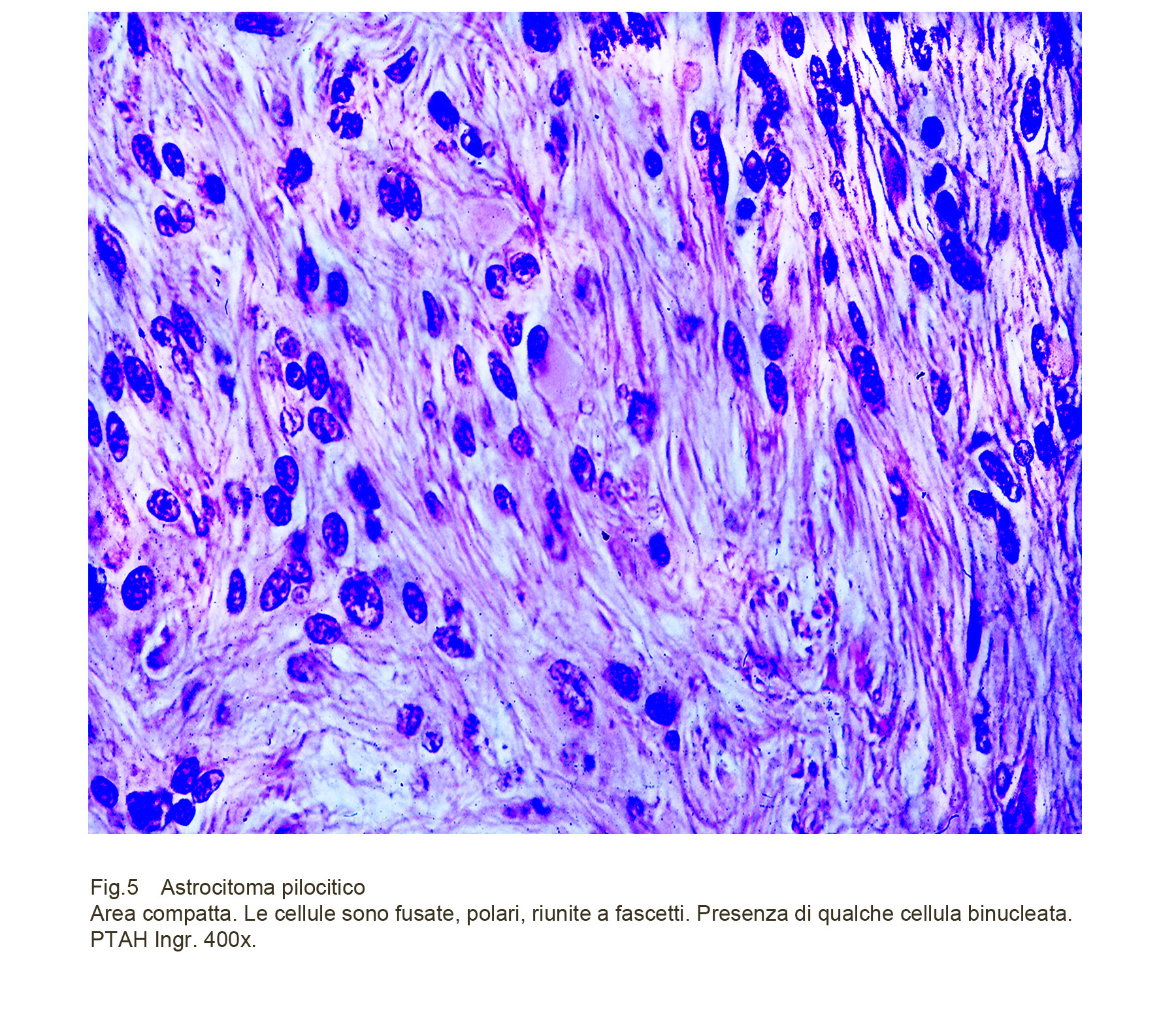
Il monomorfismo cellulare dominante può essere arricchito dalla presenza di elementi bi-nucleati, plurinucleati o irregolari oppure di forma rotondeggiante. Le mitosi e le atipie nucleari sono rare a conferma del basso indice di proliferazione di questa neoplasia; tuttavia, nei casi in cui fossero frequenti o numerose il giudizio diagnostico-prognostico non deve essere modificato.
Fig.6 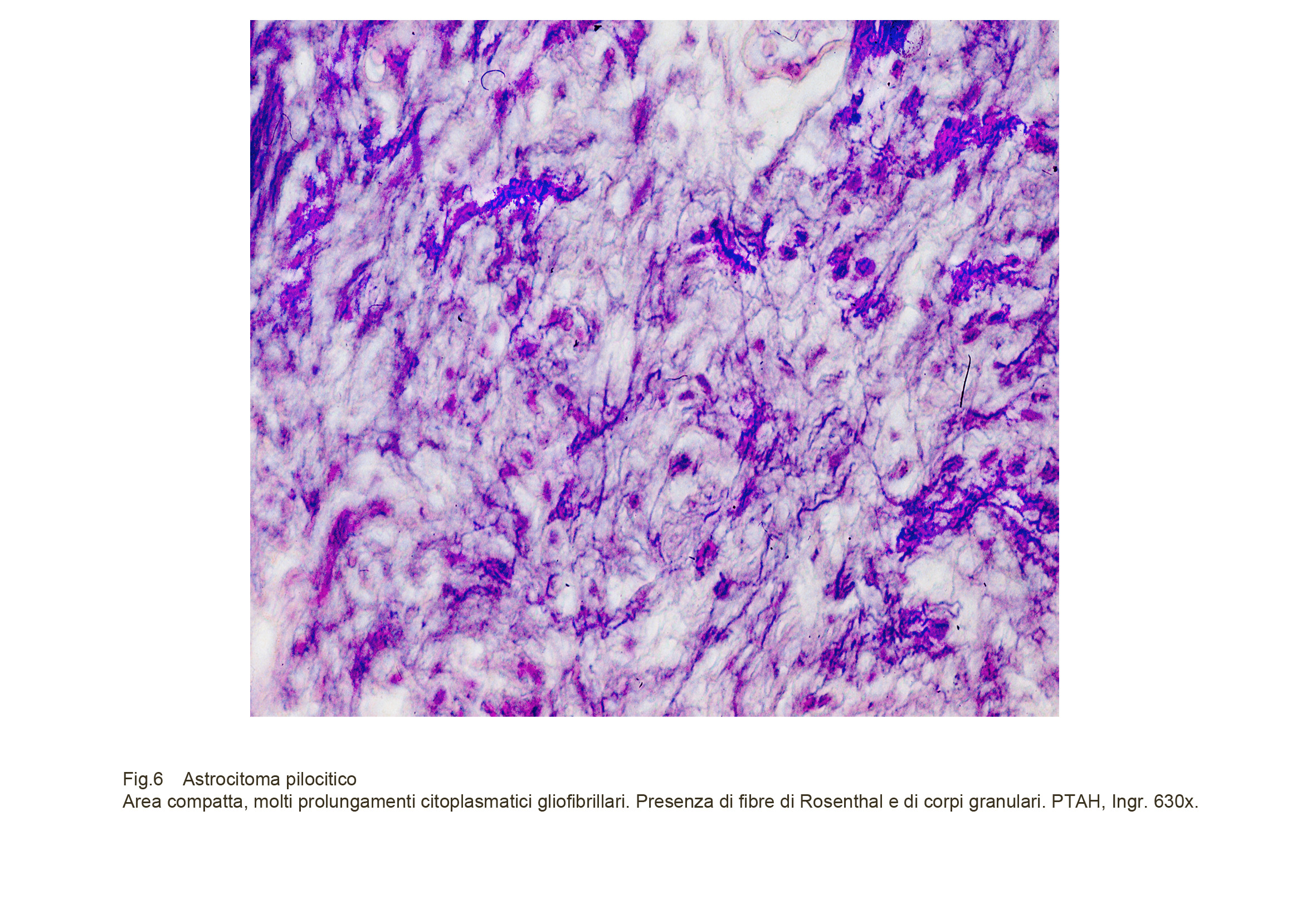
Il quadro istopatologico si rivela diverso nelle aree spongiosiche; in esse le cellule non solo sono ridotte di numero e sono disperse in ampi spazi apparentemente privi di contenuto ma evidenziano caratteri pleomorfi; infatti si repertano oltre a cellule fusate, elementi di aspetto stellato, irregolare o rotondeggiante simile ad astrociti protoplasmatici (astrocitoma pilo-protoplasmatico).
Fig.7 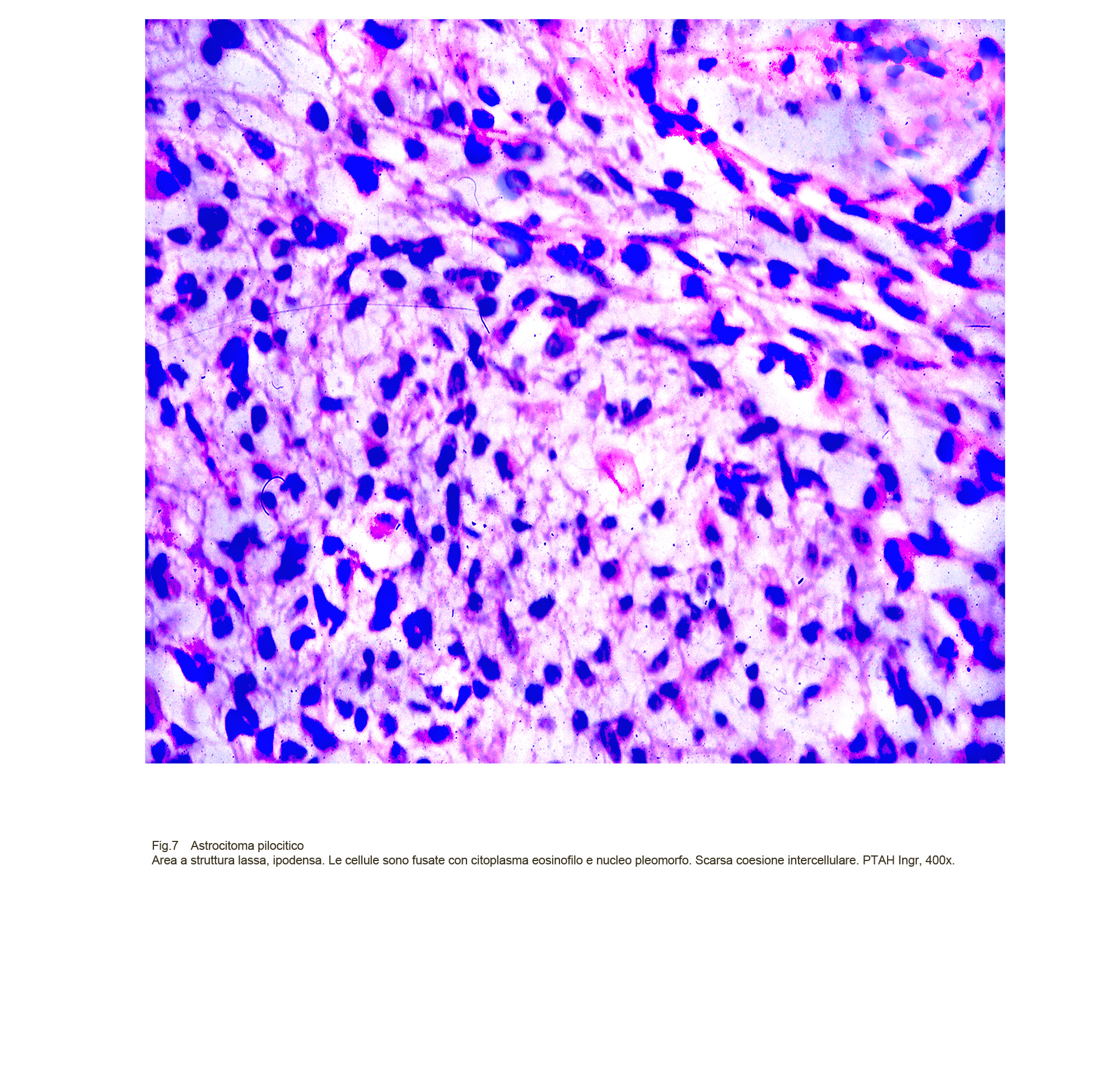
L’astrocitoma pilocitico è fornito di una rete vascolare ben sviluppata; essa è formata da capillari e da vasi di piccolo calibro. I capillari hanno pareti ben strutturate e sono rivestiti da endoteli ipertrofici o in attività proliferativa; i piccoli vasi hanno quasi sempre pareti ispessite per processi di sclerosi ma soprattutto per depositi di materiale ialino; questi vasi sono frequentemente avvolti da filiere sovrapposte di elementi pilocitici oppure sono circondati a manicotto da aggregati di cellule immunocompetenti; non è raro l’instaurarsidi processi trombiotici con conseguenti focolai di necrosi.
Fig.8 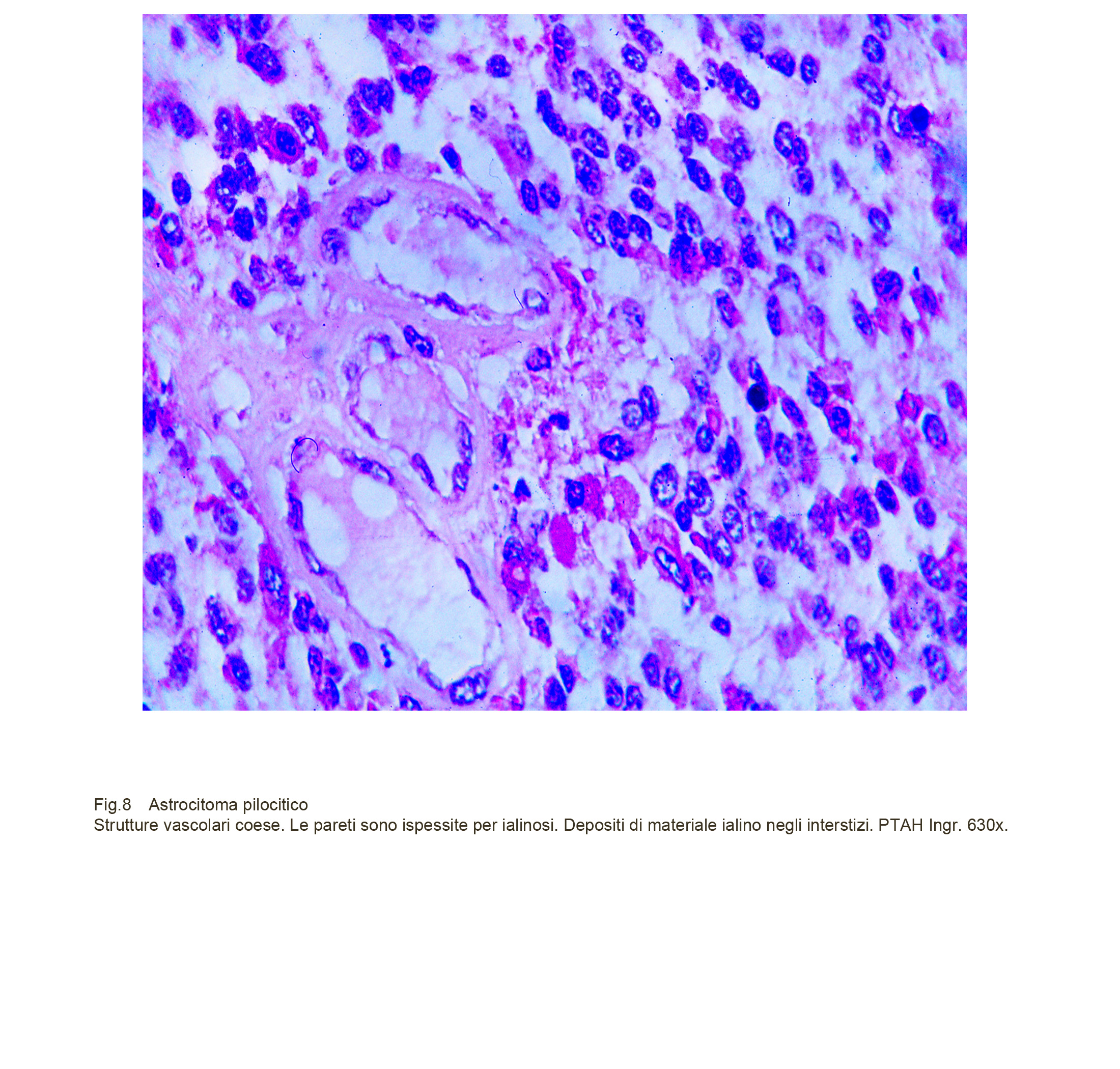
Si possono riscontrare focolai angio-proliferativi aggregati nello stesso campo microscopico fornendo immagini glomeruloidi, pseudo-angiomatoidi con lume ectasico. Un reperto microscopico caratterizzante anche se non patognomico, degli astrocitomi pilocitici è dato dalla presenza di fibre di Rosenthal e dei granuli eosinofili. Le fibre di Rosenthal appaiono come piccole formazioni cilidriche, eosinofile, compatte, omogenee della lunghezza variabile di alcuni micron; esse, prive di una struttura interna si distribuiscono in modo casuale negli spazi intercellulari e nella matrice citoplasmatica. I granuli eosinofili sono di piccole dimensioni, hanno forma irregolare, sidistribuiscono in modo isolato o sono aggregati in piccoli gruppi, hanno un aspetto compatto ma sono privi di una struttura interna; la loro densità è variabile, e si ritrovano in modo disordinato sia negli spazi intercellulari sia nel citoplasma degli elementi neoplastici.
Fig.9 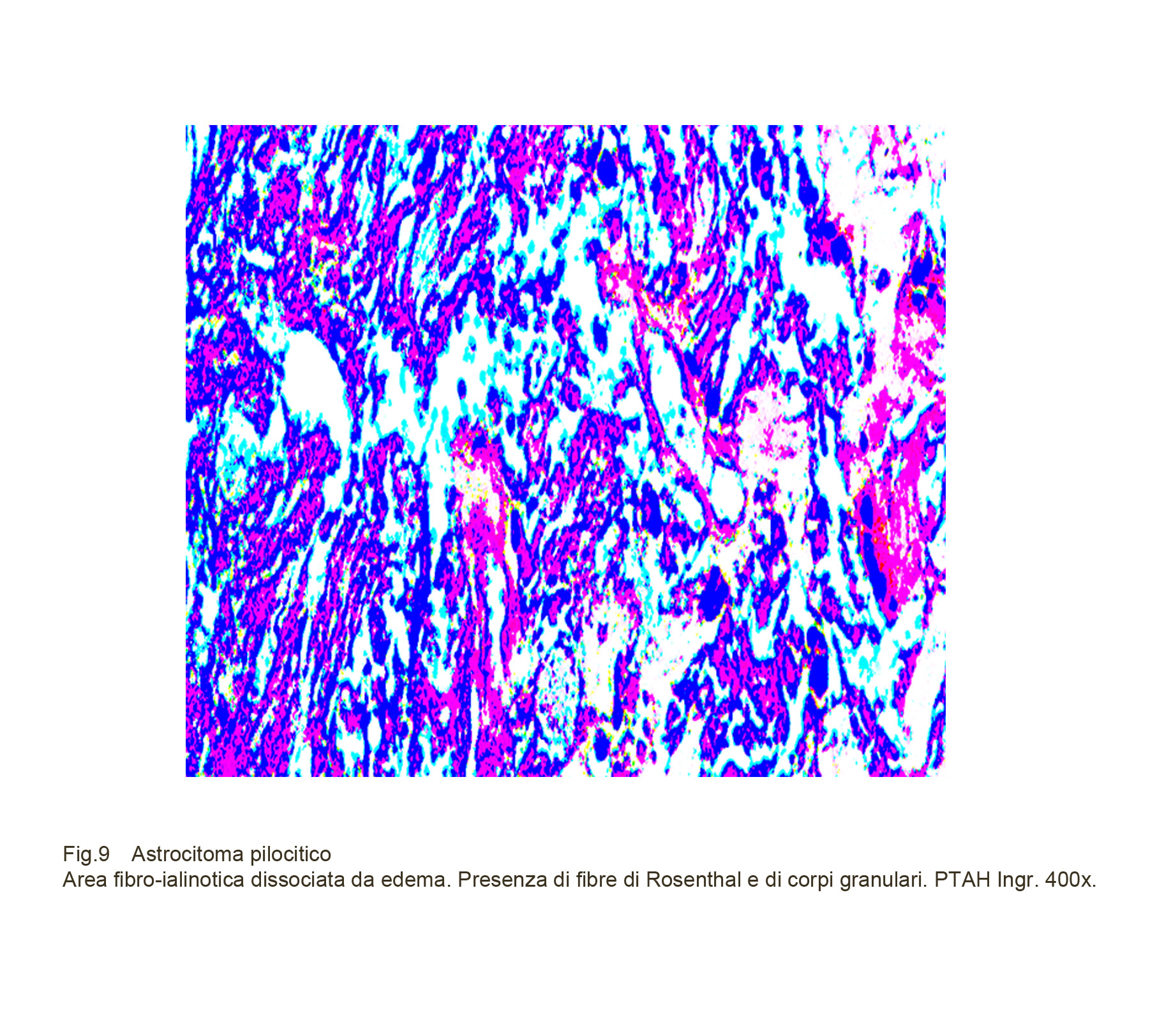
Queste due componenti sono considerate quali espressioni di processi regressivi affioranti nell’interno delle cellule e successivamente riversate negli spazi intercellulari; esse sono state repertate con maggiore densità in casi di astrocitoma pilocitico giovanile. Si sostiene che le fibre di Rosenthal siano derivate da aggregazione di materiale proteico giovanile conseguente a processi di denaturazione e alterazione chimica delle gliofibrille. Nell’ambito di questi processi regressivi deve essere sottolineata la presenza di depositi di materiale ialino; esso, in quantità variabili da caso a caso, si ritrova non solo a livello della rete vascolare e dello stroma perivascolare ma anche a livello delle cellule e degli spazi intercellulari. Allorquando interessa le sedi intra ed extracellulari, questo materiale si raccoglie sotto forma di gocce o di piccoli depositi informi.
L’astrocitoma pilocitico è frequentemente sede di processi regressivi secondari; questi, se sono numerosi e molto estesi modificano la fisionomia macroscopica e microscopica del tumore. Questi possono riguardare le cellule inducendo vacuolizzazioni; rigonfiamenti, frammentazione dei prolungamenti citoplasmatici, citolisi; possono interessare i vasi come ialinosi, sclerosi, flogosi, trombosi, emorragie, necrosi post-trombotiche; possono colpire gli spazi intercellulari e lo stroma per depositi di asli di calcio, di emosiderina o per formazione di processi antofagici e per la infiltrazione di elementi immunocompetenti.
Fig.10 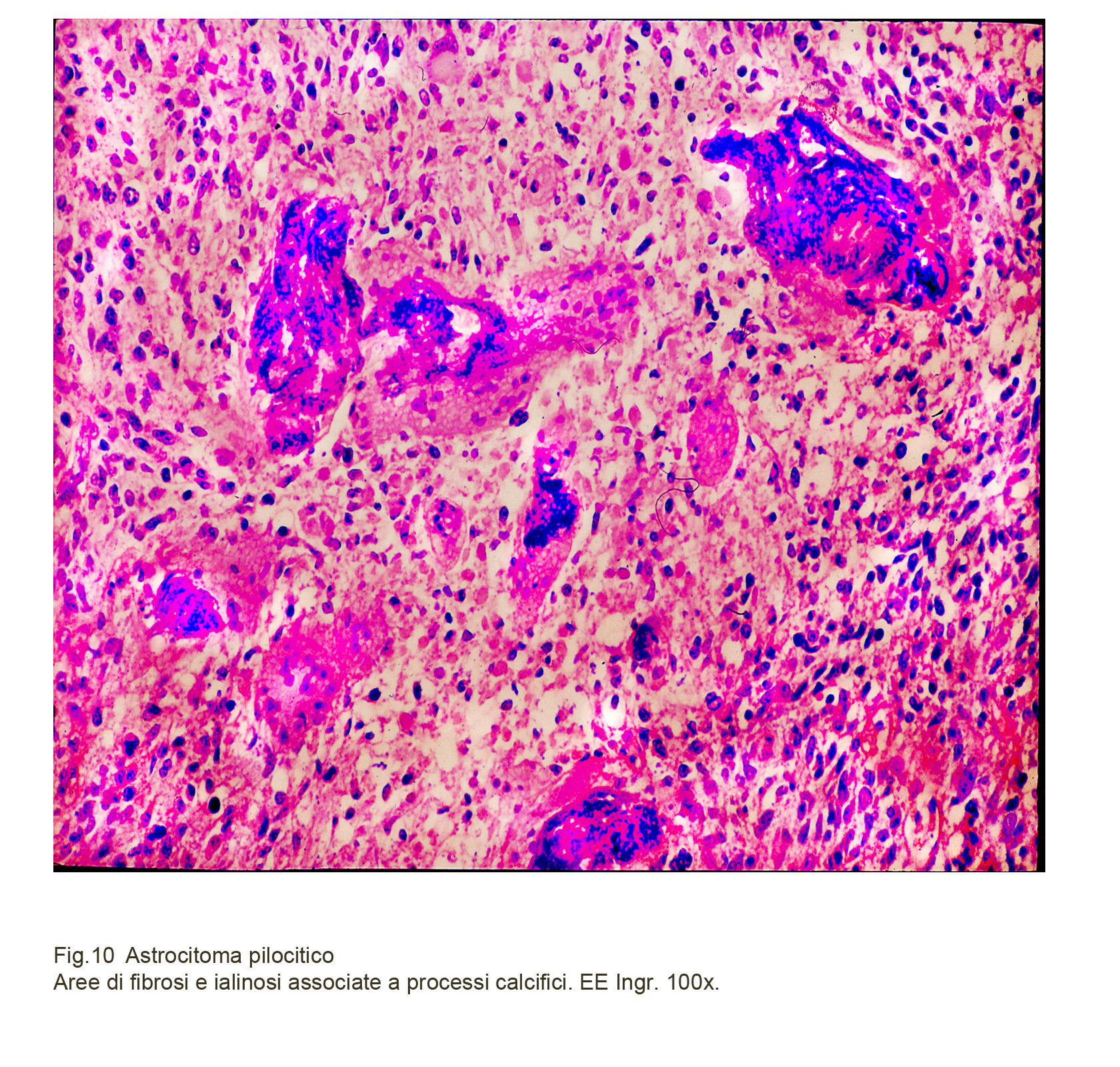
Questi processi secondari si riflettono negativamente su la prognosi e sui tempi di sopravvivenza, spesso aggravata dai seguenti mitogeni: MAPK, CREB, TOR (Acta Neuropathol., 2009, 117, 657-65). Sono descritti casi di astrocitoma pilocitico nei quali la popolazione cellulare non è diffusamente monomorfa, fusata, bipolare; invece, questi casi sono composti da una doppia o tripla popolazione cellulare astrogliale anche con aspetti morfologici transizionali. Si repertano, infatti, in associazione alla componente di base di tipo pilocitico quote variabili e plurifocali di astrociti protoplasmatici o genistocitici o di elementi riferibili a oligodendrociti. (J. Clin. Neurosc., 2011, 18, 705-709).
Questa componente astrocitaria etergenea si reperta con maggiore frequenza in casi con dominanza degli aspetti spongiosici-lassi di soggetti giovani.
IMMUNOISTOCHIMICA
L’astrocitoma pilocitico è definito, mediante l’espressività immunologica, in modo preciso sia nei suoi aspetti istogenetici sia nel suo indice di attività proliferativa. La natura astrogliale di questo tumore è svelata dalla diffusa e intensa immunoespressività per la GFAP, l’S-100, la Vimentina. L’attività proliferativa può essere determinata mediante il Ki-67, MIB-1, ottenendo dati precisi sia in senso globale sia in rapporto a poli di accrescimento o fasi transizionali in senso maligno.
Fig.11 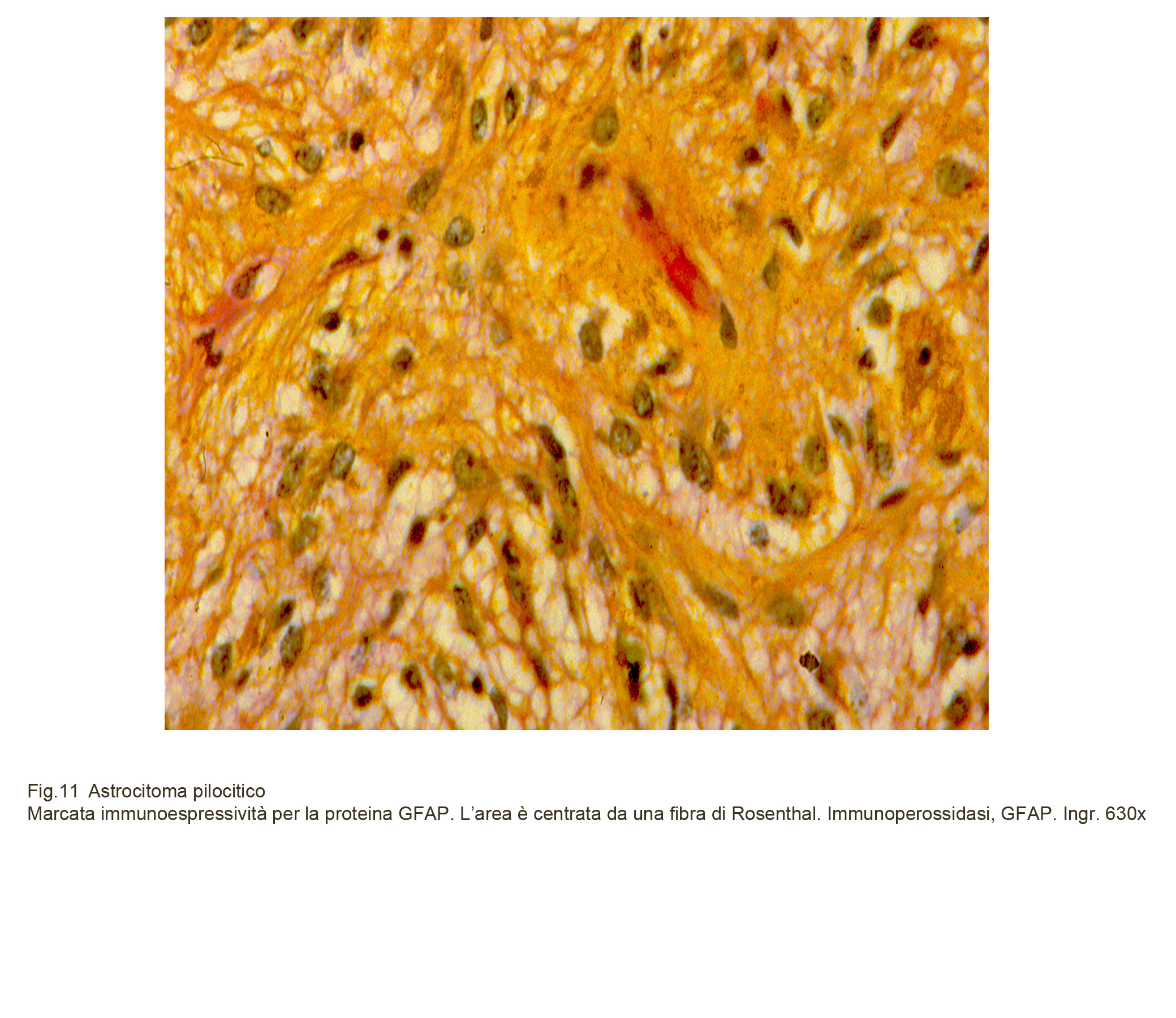
Ultrastruttura
Le cellule dell’astrocitoma pilocitico, al microscopio elettronico, evidenziano una struttura sovrapponibile a quella degli astrocitomi diffusi. Il loro citoplasma è fornito di organuli ed è sede di gliofibrille che si proiettano in sottili fasci nei prolungamenti citoplasmatici.
Fig.12 
Unico reperto da sottolineare è dato dalla presenza delle fibre di Rosenthal e dei granuli eosinofili; questo materiale al microscopio elettronico appare come aggregati densi, elettropaco, confermando così l’assenza di una propria ultrastruttura interna e la loro natura regressiva.
Fig.13 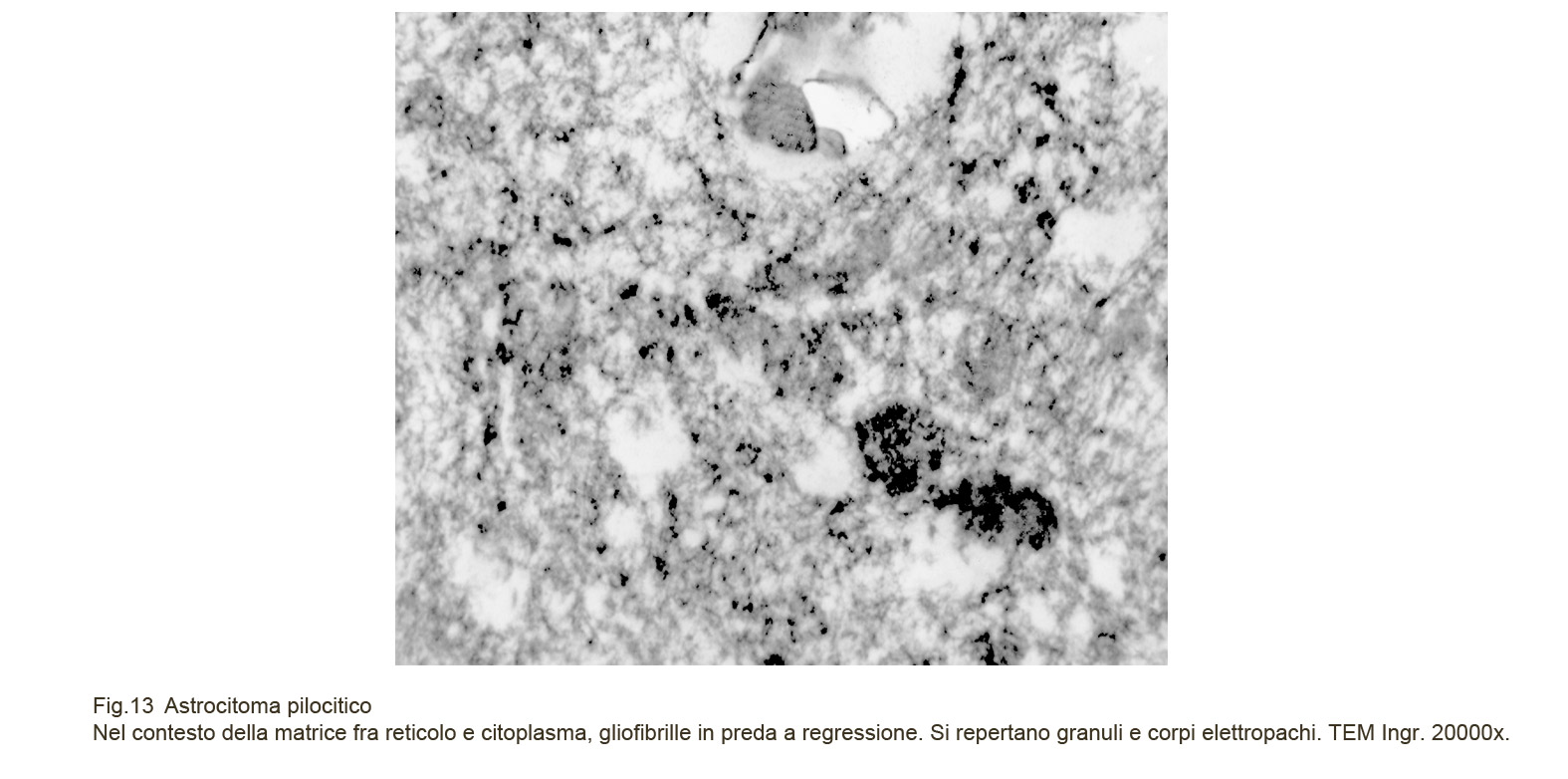 Fig.14
Fig.14 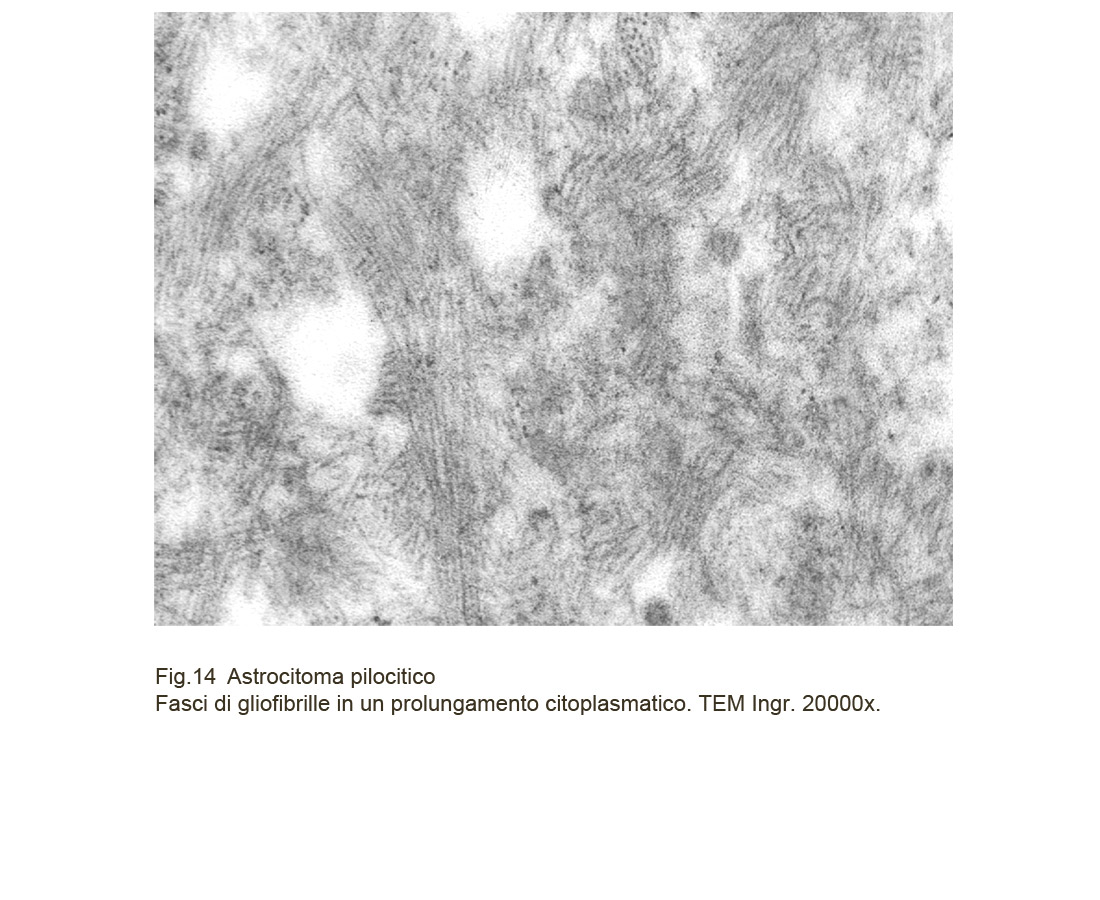 Fig.15
Fig.15 
GENETICA
L’assetto genetico-molecolare dell’astrocitoma pilocitico secondo il WHO 2007 si svolge secondo i seguenti punti di riferimento:
Perdita di PTEN
Perdita dell’eterorigosità del cromosoma 10
Delezione omozigote del gene p16
Aberrazione del gene NF1
Incremento dei livelli di galactina 3
Iperespressione del gene EF-1 a 2
Iperespressione di Erb 3 con SOX3
Mutazione di TP53
Incremento di PDGFA e PDGFRa
In letteratura sono stati riportati altri dati acquisiti e tematizzati e tra i tanti sono qui di seguito ricordati alcuni:
Duplicazione di A7q34
Iperdeterminazione della MAP-Kinasi
Fusione dei geni KJAA1549 e BRAF
Polisomia dei cromosomi 5, 6, 7, 11, 15, 20
(Acta Neuropathol. 2015, 129, 775-788)
Aumento dei cromosomi 5, 6, 7, perdita dei cromosomi 16, 17, 19, 22.
Incremento del locus 7q34 e di BRAF-MEK-ERK
(J. Neuropathology, Exp. Neurology, 2008, 67, 878-87)
Oltre alla duplicazione del cromosoma 7q34 e alla fusione dei geni KIAA/549-BRAF (fusione quale meccanismo più comune per la diagnosi genetica dell’astrocitoma pilocitico) si richiama anche l’amplificazione e il riarrangiamento dei geni HIPK2 e BRAF.
(Neurology 2009, 73, 1526-31)
(Acta Neuropathol., 2011, 121, 763-774)
(Acta Neuropathol., 2015, 129, 775-788)
Nei casi di astrocitoma pilocitico sporadico si ha un aumento del cromosoma 7q34 e un incremento del segnale BRAF-MEK-ERK (J. Neuropathology Experimental Neurology 2008, 67, 878-87).
E’ stato segnalato che l’astrocitoma pilocitico dell’adulto ha una proprietà genetica specifica rappresentata dalla mutazione dei geni JDH1 e JDH2. (Acta Neuropathol. 2009, 118, 401-05). E’ stata anche accertata una linea genetica di netta separazione tra l’astrocitoma pilocitico sporadico e quello associato a neurofibromatosi tipo 1.
Quello sporadico conserva la espressività del gene NF1 e manifesta una iperespressività del gene BRAF. L’altro (l’astrocitoma pilocitico associato a neurofibromatosi) perde la espressività dell’allele NF1 in ambedue iperespressività del gene MAPK/ERK.
(Neuropathol. Applied Neurobiology, 2000, 26, 361-67)
(J. Neuropathol. Experimental Neurology 2001, 60, 917-920)
(Histol. Histopathol. 2014, 29, 1235-48)
Gli astrocitomi pilocitici associati a neurofibromatosi tipo 1 esprimono una fusione dei geni KJAA/549 – BRAF.
(J. Pediatric. Neurosciences, 2013, 243-46)
Mediante le indagini genetiche è possibile effettuare una diagnostica che possa differenziare la forma pilocitica degli altri glioni. Infatti in essa si ritrova una iperespressività dei geni HLA-DRA, BRAF, JDH1 non repertabile negli altri tumori gliali, l’anormalità dell’attivatore mitogeno protein-Kinasi MAPK.
(J. Neuropathol. Experimental Neurology, 2005, 64, 891, 901) ( Acta Neuropathol. 2015, 129, 775-88).
E’ stato documentato che il grado di aggressività bio-morfologica è svelato anche dalle indagini genetiche; infatti gli astrocitomi pilocitici aventi caratteri di atipia sono più frequentemente sede di fusione dei geni BRAF-KIAA1549 e sotto espressione del gene ACDHILI. (J. Neurooncol. 2013, 115, 477-86)
(J. Neuropathol. Exp. Neurology 2008, 67, 1194-1204).
Le ricerche genetiche hanno esplorato anche la possibilità di prevedere l’arco prognostico di sopravvivenza mediante la valutazione del grado di mutazione dell’istoni H33K27M e GB4V/B. (Ann. Clin. Transl. Neurol. 2015, 2, 439-43).
Astrocitoma pilomixoide (PMA)
Definizione
L’astrocitoma pilomixoide (PMA) è una variante dell’astrocitoma pilocitico; esso è formato da cellule fusate, monomorfe, aventi come carattere dominante una disposizione spaziale angiocentrica con ampi spazi intercellulari occupati da materiale mucoide.
Sede
L’astrocitoma pilomixoide si ritrova di prevalenza a livello della zona ipotalamo-chiasmatica; con minore frequenza è sede a livello del talamo, del cervelletto, del ponte, dei lobi temporali, del midollo spinale.
Caratteri macroscopici
La neoplasia mostra nell’insieme un aspetto gelatinoso; infatti è di colore bianco-giallastro, ha una consistenza molle e in alcuni casi diffluente; si taglia con difficoltà e le superfici di sezione sono compatte o finemente spongiosiche, non mantengono i bordi del taglio e mostrano margini sfusati o indistinguibili rispetto ai tessuti circostanti.
Caratteri istopatologici
L’immagine microscopica d’insieme evidenzia due aspetti determinanti; il primo è rappresentato dalla preponderante aggregazione delle cellule attorno ai piccoli vasi. Questi sono numerosi, sono distribuiti in modo disordinato e mostrano tutti pareti ispessite per processi di fibrosi con slaminamento degli strati fibrillari concentrici; il rivestimento endoteliale è ipertrofico, sporge nel lume e a livello dei capillari, mostra focolai di attività proliferativa. Questi capillari nel loro manifestarsi secondo modalità iperplastico-proliferativa acquisiscono frequentemente aspetti glomeruloidi.
Il secondo è dato dagli ampi spazi occupati da metariale amorfo, poco tingibile, colorabile con i metodi per la mucina.
Fig.16 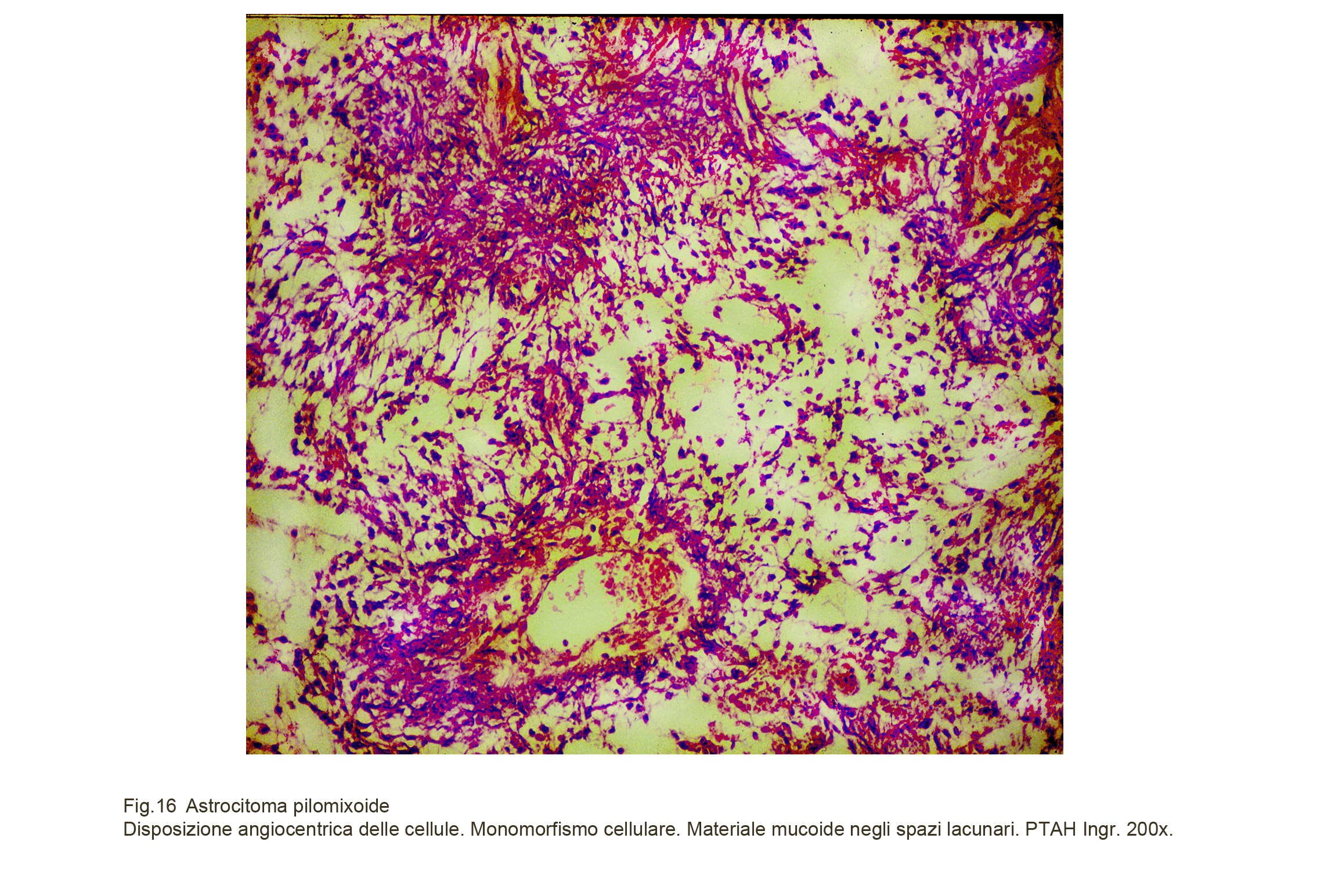
A un esame istopatologico analitico e quindi dettagliato si rileva che la popolazione cellulare è isomorfa ed è costituita da cellule fusate, digitiformi, bipolari, con lunghi prolungamenti citoplasmatici che si staccano dai poli. Questi elementi hanno una esigua quota citoplasmatica che avvolge un nucleo oblungo, normocromatinico e nucleolato; frequentemente queste cellule sono in mitosi e i loro nuclei sono irregolari, ipercromatinici, voluminosi, atipici. La disposizione spaziale di queste cellule è particolare poiché esse in prevalenza si aggregano, creando immagini di cespugli iperdensi, attorno ai piccoli vasi. Quasi in contrasto quote esigue delle stesse cellule sono distribuite in ampi spazi intercellulari. Queste si dispongono anche in modo da formare pseudorosette o rosette vere come negli ependimomi (J. Pediatric. Neuroscience, 2013, 8, 243-46).
Fig.17 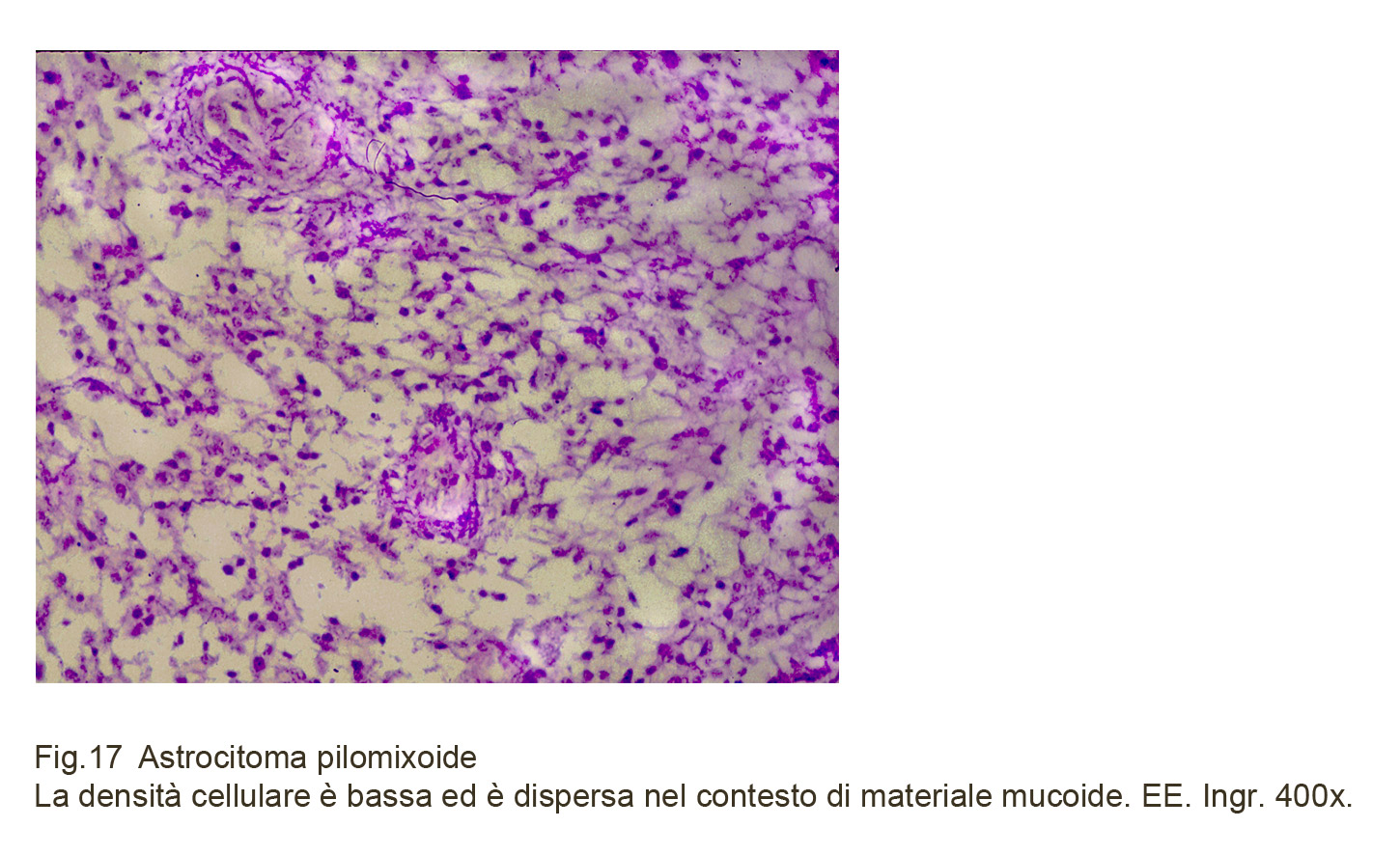
In nessun campo microscopico sono rilevabili fibre di Rosenthal e granuli eosinofili e gocce ialine. Questi spazi, con aspetto lacunare sono occupati da materiale amorfo, tingibile, di natura mucoide.
Sono molto frequenti i processi regressivi secondari; tra di essi sono da ricordare la presenza di cellule immunocompetenti raccolte intorno ai vasi e la esistenza di focolai di necrosi coagulativa da microtrombi.
Immunoistochimica
Il quadro di espressività immunologiche conferma la natura astrogliale di questa neoplasia e sottolinea un indice di attività proliferativa rispetto all’astrocitoma pilocitico. La natura astrogliale viene accertata attraverso la immunopositività diffusa per la GFAP, la S-100, La Vimentina; l’attività proliferativa viene letta mediante un’alta espressività immunologica per il Ki-67.
Fig.18 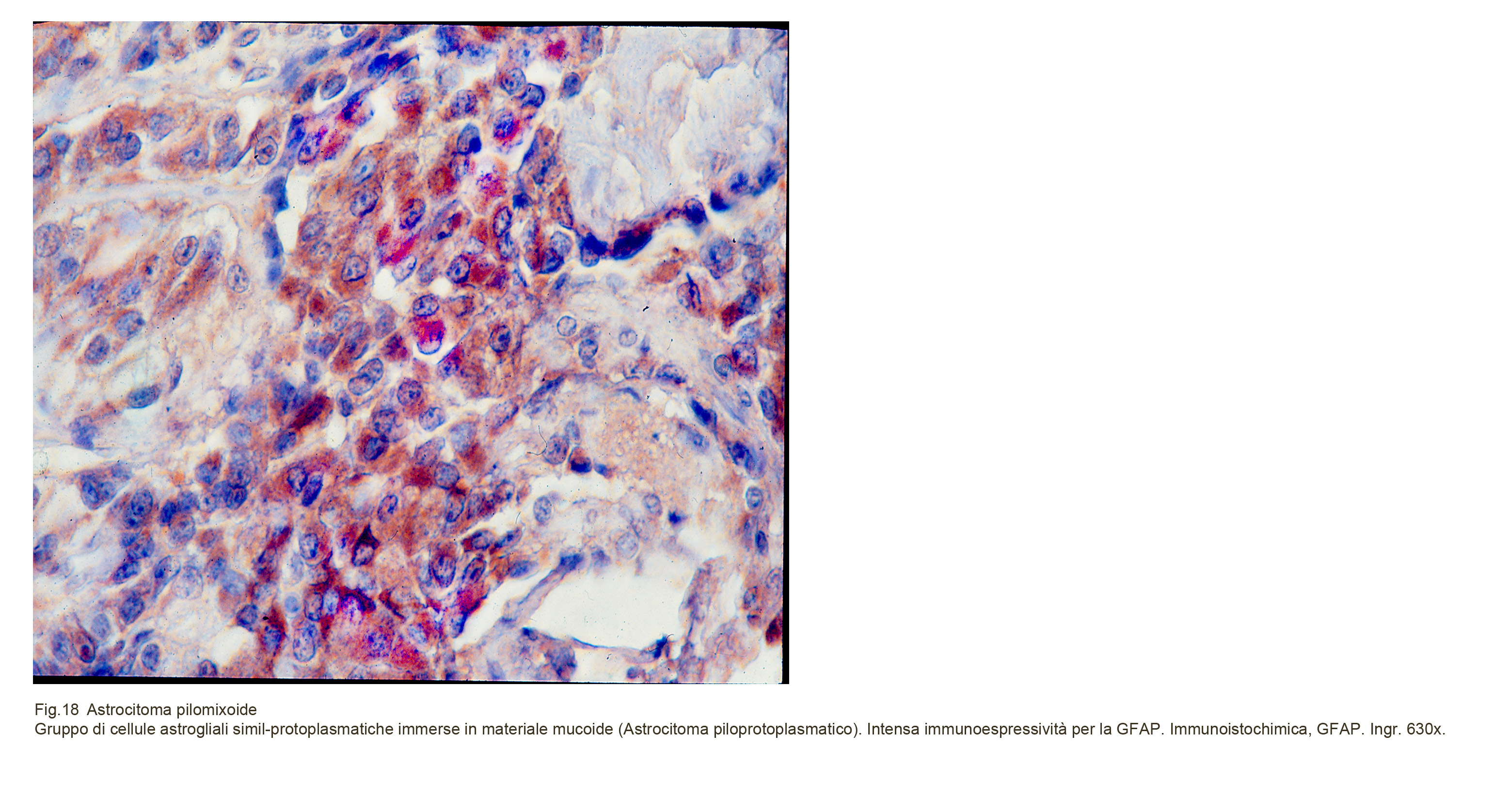
(J. Pediatric Neurosciences, 2013, 8, 243-46) (J. Clin. Neurosc. 2014, 21, 1993-6).
GENETICA
L’astrocitoma pilomixoide in quanto variante dell’astrocitoma pilocitico svela la propria autonomia nosografica anche a livello del proprio assetto genetico e del proprio indice proliferativo. E’ stato documentato un ipersviluppo dei geni H-19, DACT2, JMP3, COL2A1, COL1A1 e una espressività elevata del MIB-1. (Surg. Neurol. Int. 2014, 5, 29-36) (Brain Pathol. 2015, 25, 429-40).
Il gene ALDHJL1 è sottoespresso nei casi di astrocitoma pilomixoide così come nelle forme di astrocitoma pilocitico aggressive o maligne. (J. Neuropathol. Exp. Neurol. 2008, 67, 1194-1204).
ASTROCITOMA PILOCITICO ANAPLASTICO
In letteratura sono segnalati e descritti casi di astrocitomi pilocitici aventi caratteri morfologici e immunoistochimici di anaplasia o quelli (più vari) del glioblastoma multiforme.
(Brain Tumor Pathol., 2014, 31, 108-12).
Questi eventi possono manifestarsi nel contesto di un preesistente astrocitoma pilocitico benigno, soprattutto nei casi in cui il paziente era stato sottoposto a radioterapia; queste possibilità sono sempre prese in considerazione e il loro manifestarsi sono indicate come astrocitomi pilocitici anaplastici secondari (Clin. Neurol. Neurosurg., 2013, 115, 1220-5).
Sono riportati anche rari casi di astrocitomi pilocitici anaplastici insorgenti come tali, mostrando fin dall’inizio caratteri bio-morfologici di malignità; questi sono etichettati quali astrocitomi pilocitici anaplastici primitivi. Le forme secondarie, più frequenti, si manifestano inizialmente in modo unifocale o plurifocale e successivamente, in conseguenza della agressività biologica della componente anaplastica, infiltrano progressivamente la pre-esistente neoplasia fino a sostituirla ed invadere le strutture vicine.
(Brain Tumor Pathol., 2014, 31, 108-12) (Childs Nerv. Syst., 2015, 31, 167-71).
Il rischio di un processo di trasformazione anaplastico è maggiore dopo trattamento radioterapico, ma esso permane anche se con minore incidenza se si rinuncia a tale trattamento terapeutico.
Fanno eccezione le forme giovanili, le quali possono subire tale trasformazione solo dopo radioterapia. (Br. J. Ophthalmol. 2008, 92, 40-6).
I dati statistici concordano nell’evidenziare una maggiore frequenza di processi anaplastici in casi di astrocitoma pilocitico sporadico rispetto a quelli associati a NF1. (J. Pediatr. Hematol. Oncol. 2011, 33, 198-201).
I caratteri istopatologici di queste forme anaplastiche sono costituiti da iperdensità cellulare, da marcato pleomorfismo, da un elevato numero di mitosi, da gravi atipie cellulari e nucleari, dalla presenza di aree di necrosi, da una marcata neoangiogenesi esplosa in modo caotico, dalla presenza di cellule mostruose bi-nucleate o pluri-nucleate; da un alto indice di attività proliferativa. (J. Clin. Neuroscience, 2014, 21, 1193-6).
La citomorfologia preponderante può essere variabile e può mostrare aspetti di transizione; in tale ambito sono stati individuati i seguenti aspetti morfologici:
- Cellule pilocitico-like
- Cellule piccole indifferenziate
- Cellule epitelioidee o rabboidee
- Cellule simili ad astrocitoma diffuso anaplastici.
Le indagini di espressività immunologiche confermano la natura astrogliale della neoplasia anaplastica per l’alta e diffusa presenza di proteina GFAP nel citoplasma delle cellule; a questo si associa una alta determinazione di positività per il Ki67 e il MIB-1, attestante un indice di attività proliferativa di grado notevole. Anche le indagini genetiche svelano uno specifico profilo che può essere così riportato:
Mutazione di TP53, PTEN, BRAF, V600E Gain del cromosoma 7.
Iperespressività del fattore EGFR
Delezione dei cromosomi 6, 9p, 11p, 19q, 22q
Amplificazione ed iperespressività dei geni CDK4, 12q, 13q1 (Indian J. Cancer, 2009, 46, 108-119)
Al suddetto quadro è necessario integrare l’attivazione dei geni mitogeni MAPK, CREB, TOR
Delezione del gene p16
(Acta Neuropathol, 2009, 117, 657-65)
(Clin. Neuropathol. , 2013, 32, 159-64)
Si ha l’attivazione dei seguenti geni: MAPK, CREB, nTOR
(Acta Neuropathol., 2009, 117, 657-65).

